
PHYSICIANS
PHYSICIAN RESOURCES
Nature Reviews Rheumatology (www.nature.com)
Annals of the Rheumatic Diseases- (www.nature.com)
Arthritis & Rheumatology- (www.onlinelibrary.wiley.com)
Arthritis Research & Therapy- (www.arthritis-research.com)
Current Opinion in Rheumatology- (www.journals.lww.com)
Current Rheumatology Reports- (www.springer.com)
Lupus- (www.lup.sagepub.com)
Rheumatology- (www.rheumatology.oxfordjournals.org)
The Journal of Rheumatology- (www.jrheum.org)
Indian journal of rheumatology- (www.indianjrheumatol.com)
Journal of Bone and Mineral Research- (www.jbmr.org)
Arthritis Care & Research- (www.onlinelibrary.wiley.com)
Seminars in Arthritis and Rheumatism- (www.semarthritisrheumatism.com )
Best practice & research in clinical rheumatology- (www.elsevier.com)
Clinical and Experimental Rheumatology- (www.clinexprheumatol.org)
Rheumatic Disease Clinics of North America- (www.sciencedirect.com)
Scandinavian Journal of Rheumatology- (www.tandfonline.com)
Modern Rheumatology- (www.springer.com)
BMC Musculoskeletal Disorders- (www.biomedcentral.com)
Clinical Rheumatology- (www.springer.com)
International Journal of Rheumatic Diseases- (www.wiley.com)
Compiled and prepared by
Dr Sundaram T G
SGPGI, Lucknow
Dr Avinash Jain
SGPGI, Lucknow
1. Rheumatoid Arthritis
2010 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Rheumatoid Arthritis:
Target population (Who should be tested?)1: Patients who
- 1. Have at least one joint with definite clinical synovitis (swelling)
- 2. With the synovitis not better explained by another disease2
Classification criteria for RA (score-based algorithm: add score of categories A–D; a score of ≥6 of 10 is needed for classification of a patient as having definite RA)3
| A. Joint involvement4 | Score |
|---|---|
| 1 large joint5 | 0 |
| 2–10 large joints | 1 |
| 1–3 small joints6 (with or without involvement of large joints) | 2 |
| 4–10 small joints (with or without involvement of large joints) | 3 |
| >10 joints7 (at least 1 small joint) | 5 |
| B. Serology (at least 1 test result is needed for classification)8 | |
| Negative RF and negative ACPA | 0 |
| Low-positive RF or low-positive ACPA | 2 |
| High-positive RF or high-positive ACPA | 3 |
| C. Acute-phase reactants (at least 1 test result is needed for classification)9 | |
| Normal CRP and normal ESR | 0 |
| Abnormal CRP or abnormal ESR | 1 |
| D. Duration of symptoms10 | |
| <6 weeks | 0 |
| ≥6 weeks | 1 |
- 1. In addition, patients with erosive disease typical of rheumatoid arthritis (RA) with a history compatible with prior fulfilment of the 2010 criteria should be classified as having RA. Patients with longstanding disease, including those whose disease is inactive (with or without treatment) who, based on retrospectively available data, have previously fulfilled the 2010 criteria should be classified as having RA.
- 2. Differential diagnoses vary among patients with different presentations but may include conditions such as systemic lupus erythematosus, psoriatic arthritis, and gout. If it is unclear about the relevant differential diagnoses to consider, an expert rheumatologist should be consulted.
- 3. Although patients with a score of 6 of 10 are not classifiable as having RA, their status can be reassessed, and the criteria might be fulfilled cumulatively over time.
- 4. Joint involvement refers to any swollen or tender joint on examination, which may be confirmed by imaging evidence of synovitis. Distal interphalangeal joints, first carpometacarpal joints, and first metatarsophalangeal joints are excluded from assessment. Categories of joint distribution are classified according to the location and number of involved joints, with placement into the highest category possible based on the pattern of joint involvement.
- 5. “Large joints” refers to shoulders, elbows, hips, knees, and ankles.
- 6. “Small joints” refers to the metacarpophalangeal joints, proximal interphalangeal joints, second through fifth metatarsophalangeal joints, thumb interphalangeal joints, and wrists.
- 7. In this category, at least one of the involved joints must be a small joint; the other joints can include any combination of large and additional small joints, as well as other joints not specifically listed elsewhere (e.g., temporomandibular, acromioclavicular, sternoclavicular).
- 8. Negative refers to IU values that are less than or equal to the upper limit of normal (ULN) for the laboratory and assay; low-positive refers to IU values that are higher than the ULN but ≤3 times the ULN for the laboratory and assay; high-positive refers to IU values that are ≥3 times the ULN for the laboratory and assay. When rheumatoid factor (RF) information is only available as positive or negative, a positive result should be scored as low positive for RF.
- 9. Normal and abnormal are determined by local laboratory standards.
- 10. Normal and abnormal are determined by local laboratory standards.
Reference: Singapore Society Rheumatology (SSR) website Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569-2581.
2. Sjögren Syndrome
2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren Syndrome:
The classification of primary Sjögren syndrome applies to any individual who meets the inclusion criteria1, does not have any of the conditions listed as exclusion criteria2,and has a score of ≥4 when the weights from the five criteria items below are summed.
| Labial salivary gland with focal lymphocytic sialadenitis and focus score of ≥1 foci/4 mm23 | 3 |
|---|---|
| Anti-SSA/Ro positive | 3 |
| Ocular staining score ≥5 (or van Bijsterveld score ≥4) in at least one eye4, 5 | 1 |
| Schirmer test ≤5 mm/5 minutes in at least one eye4 | 1 |
| Unstimulated whole saliva flow rate ≤0.1 mL/min6 | 1 |
1. These inclusion criteria are applicable to any patient with at least one symptom of ocular or oral dryness, defined as a positive response to at least one of the following questions:
- Have you had daily, persistent, troublesome dry eyes for more than 3 months?
- Do you have a recurrent sensation of sand or gravel in the eyes?
- Do you use tear substitutes more than three times a day?
- Have you had a daily feeling of dry mouth for more than 3 months?
- Do you frequently drink liquids to aid in swallowing dry food?
or in whom there is suspicion of Sjögren syndrome (SS) from the European League Against Rheumatism SS Disease Activity Index questionnaire (at least one domain with a positive item).
2. Exclusion criteria include prior diagnosis of any of the following conditions, which would exclude diagnosis of SS and participation in SS studies or therapeutic trials because of overlapping clinical features or interference with criteria tests: (1) history of head and neck radiation treatment, (2) active hepatitis C infection (with confirmation by PCR), (3) AIDS, (4) sarcoidosis, (5) amyloidosis, (6) graft-versus-host disease, or (7) IgG4-related disease.
3. The histopathologic examination should be performed by a pathologist with expertise in the diagnosis of focal lymphocytic sialadenitis and focus score count using the protocol described by Daniels et al.
4. Patients who are normally taking anticholinergic drugs should be evaluated for objective signs of salivary hypofunction and ocular dryness after a sufficient interval without these medications for these components to be a valid measure of oral and ocular dryness.
5. Ocular Staining Score described by Whitcher et al; van Bijsterveld score described by van Bijsterveld.
6. Unstimulated whole saliva flow rate measurement described by Navazesh and Kumar.
Reference: Shiboski CH, Shiboski SC, Seror R, et al; International Sjögren’s Syndrome Criteria Working Group. 2016 American College of Rheumatology/European League Against Rheumatism Classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheum. 2017;69(1):35-45.
2012 American College of Rheumatology Proposed Classification Criteria for Sjögren Syndrome1
The classification of Sjögren syndrome (SS), which applies to individuals with signs or symptoms that may be suggestive of SS, will be met in patients who have at least two of the following three objective features:
- 1. Positive serum anti-SSA/Ro and/or anti-SSB/La or (positive RF and ANA titer ≥ 1:320)
- 2. Labial salivary gland biopsy exhibiting focal lymphocytic sialadenitis with a focus score ≥1 focus/4 mm2 2
- 3. Keratoconjunctivitis sicca with ocular staining score ≥3 (assuming that individual is not currently using daily eye drops for glaucoma and has not had corneal surgery or cosmetic eyelid surgery in the past 5 years)3
Prior diagnosis of any of the following conditions would exclude participation in SS studies or therapeutic trials because of overlapping clinical features or interference with criteria tests:
History of head and neck radiation treatment
Hepatitis C infection
Acquired immunodeficiency syndrome
Sarcoidosis
Amyloidosis
Graft-versus-host disease
IgG4-related disease
- We excluded participants with rheumatoid arthritis, systemic lupus erythematosus, scleroderma, and other connective tissue disease from these analyses because there were only 87 (6%) such participants.
- Using histopathologic definitions and focus score assessment methods as described by Daniels et al.
- Using ocular staining score as described by Whitcher et al.
References: Shiboski SC, Shiboski CH, Criswell LA, et al. American College of Rheumatology Classification Criteria for Sjögren’s Syndrome: a data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance Cohort. Arthritis Care Res. 2012;64(4):475-487.
Whitcher, John P., Caroline H. Shiboski, Stephen C. Shiboski, Ana Maria Heidenreich, Kazuko Kitagawa, Shunhua Zhang, and Steffen Hamann et al. 2010. "A Simplified Quantitative Method For Assessing Keratoconjunctivitis Sicca From The Sjögren's Syndrome International Registry". American Journal Of Ophthalmology 149 (3): 405-415.
Revised International Classification Criteria for Sjogren’s syndrome (2002)
| Symptoms | Duration/Score |
|---|---|
| I. Ocular symptoms: a positive response to at least one of the following questions: | ≥ 3 months |
| 1. Have you had daily, persistent, troublesome dry eyes for more than 3 months? | - |
| 2. Do you have a recurrent sensation of sand or gravel in the eyes? | - |
| 3. Do you use tear substitutes more than 3 times a day? | ≥ 3/day |
| II. Oral symptoms: a positive response to at least one of the following questions: | ≥ 3 months |
| 1. Have you had a daily feeling of dry mouth? | - |
| 2. Have you had recurrently or persistently swollen salivary glands as an adult? | - |
| 3. Do you frequently drink liquids to aid in swallowing dry food? | - |
| III. Ocular signs—that is, objective evidence of ocular involvement defined as a positive result for at least one of the following two tests: | |
| 1. Schirmer’s I test, performed without anaesthesia | ≤5 mm in 5 minutes |
| 2. Rose Bengal score or other ocular dye score | ≥4 according to van Bijsterveld’s scoring system |
| IV. Histopathology | |
| In minor salivary glands (obtained through normal-appearing mucosa), focal lymphocytic sialadenitis, evaluated by anexpert histopathologist, with a focus score greater than 1, defined as a number of lymphocytic foci (which are adjacent to normal-appearingmucous acini and contain more than 50 lymphocytes) per 4 mm2 of glandular tissue | FLS ≥ 1 |
| V. Salivary gland involvement: objective evidence of salivary gland involvement defined by a positive result for at least one of the following diagnostic tests: | |
| 1. Unstimulated whole salivary flow | ≤1.5 mL in 15 minutes |
| 2. Parotid sialography showing the presence of diffuse sialectasias (punctate, cavitary or destructive pattern), without evidence of obstruction in the major ducts | - |
| 3. Salivary scintigraphy showing delayed uptake, reduced concentration, and/or delayed excretion of tracer | - |
| VI. Autoantibodies: | |
| Presence in the serum of the following autoantibodies: Antibodies to Ro(SSA) or La(SSB) antigens, or both |
Revised Rules for Classification
For Primary SS
In patients without any potentially associated disease, primary SS may be defined as follows:
- A. The presence of any 4 of the 6 items is indicative of primary SS, as long as either item IV (Histopathology) or VI (Serology) is positive.
- B. Any 3 of the 4 objective criteria items (i.e., items III, IV, V, and VI) are present.
- C. The classification tree procedure represents a valid alternative method for classification, although it should be more properly used in clinical-epidemiologic surveys.
For Secondary SS
In patients with a potentially associated disease (for instance, another well-defined connective tissue disease), the presence of item I or item II plus any 2 from among items III, IV, and V may be considered as indicative of secondary SS.
Exclusion Criteria
- Past head and neck radiation treatment
- Hepatitis C infection
- Acquired immunodeficiency syndrome
- Pre-existing lymphoma
- Sarcoidosis
- Graft-versus-host disease
- Use of anticholinergic drugs (since a time shorter than fourfold the half-life of the drug)
Reference: Vitali C, Bombardieri S, Jonsson R, et al and the European Study Group on Classification Criteria for Sjögren’s Syndrome, Ann Rheum Dis 61:554–558, 2002
3. Spondyloarthritis
Axial:
ASAS classification criteria for axial SpA
Inclusion - Patients with back pain- 3 months and age at onset <45 years
with
Sacroiliitis on imaging* plus 1 SpA feature**
(or)
HLA-B27 plus 2 other SpA features**
* Sacroiliitis on imaging:
Active (acute) inflammation on MRI highly suggestive of sacroiliitis associated with SpA
or
Definite radiographic sacroiliitis according to modified New York criteria
(B/L grade ³ 2 Sacroiliitis or U/L Grade ³ 3 Sacroiliitis
** SpA features:
- Inflammatory back pain
- Arthritis
- Enthesitis (heel)
- Uveitis
- Dactylitis
- Psoriasis
- Crohn’s disease/ulcerative colitis
- Good response to NSAIDs
- Family history for SpA
- HLA-B27
- Elevated CRP
Specification of the Variables Used for the Assessment of SpondyloArthritis International Society Criteria for Classification of Axial Spondyloarthritis:
Inflammatory Back Pain (IBP) - IBP according to experts: four out of five of the following parameters present: (1) age at onset <40 years, (2) insidious onset, (3) improvement with exercise, (4) no improvement with rest, or (5) pain at night (with improvement upon getting up)
Arthritis - Past or present active synovitis diagnosed by a doctor
Family history - Presence in first- or second-degree relatives of any of the following: (a) ankylosing spondylitis, (b) psoriasis, (c) uveitis, (d) reactive arthritis, or (e) inflammatory bowel disease
Psoriasis - Past or present psoriasis diagnosed by a doctor
Inflammatory bowel disease - Past or present Crohn’s Disease or Ulcerative Colitis diagnosed by adoctor
Dactylitis - Past or present dactylitis diagnosed by a doctor
Enthesitis - Heel enthesitis: past or present spontaneous pain or tenderness at examination at the site of the insertion of the Achilles tendon or plantar fascia at the calcaneus
Anterior Uveitis - Past or present uveitis anterior confirmed by an ophthalmologist
Good response to NSAIDs - At 24–48 hours after a full dose of NSAID, the back pain is not present anymore or is much better
HLA-B27 - Positive testing according to standard laboratory techniques
Elevated CRP - CRP above upper normal limit in the presence of back pain after exclusion of other causes for elevated CRP concentration
Sacroiliitis by radiography - Bilateral grade 2–4 or unilateral grade 3–4 according to the modified New York criteria
Sacroiliitis by MRI - Active inflammatory lesions of sacroiliac joints with definite bone marrow edema or osteitis suggestive of sacroiliitis associated with SpA
Reference: Sieper J, Rudwaleit M, Baraliakos X, et al. The Assessment of SpondyloArthritis International Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis. 2009;68(suppl 2):ii1-44.
Comparison of Various Criteria for the Diagnosis of Inflammatory Back Pain
IBP according to ASAS experts1 to be applied in patients with chronic back pain (>3 months)
| ASAS1 | Calin2 | Berlin3 | |
|---|---|---|---|
| Age (in years) | < 40 | < 40 | |
| Duration (in months) | > 3 | > 3 | |
| Onset | Insidious | Insidious | |
| Worsening | Night | Awakening second half of night | |
| Improvement | Exercise On getting up at night NO improvement with rest | Exercise | Exercise but not with rest |
| Associated features | Early Morning stiffness (EMS) | Alternating buttock pain EMS > 30 min |
- 1. Sieper J, van der Heijde D, Landewé R, et al. New criteria for inflammatory back pain in patients with chronic back pain: a real patient exercise by experts from the Assessment of SpondyloArthritis International Society (ASAS). Ann Rheum Dis. Jun;68(6):784-788.
- 2. Calin A, Porta J, Fries JF, Schurman DJ. Clinical history as a screening test for ankylosing spondylitis. JAMA. 1977;237(24):2613-2614.
- 3. Rudwaleit M, Metter A, Listing J, et al. Inflammatory back pain in ankylosing spondylitis: a reassessment of the clinical history for application as classification and diagnostic criteria. Arthritis Rheum. 2006;54(2):569-578.
Reference:Sieper J, Rudwaleit M, Baraliakos X, et al. The Assessment of SpondyloArthritis International Society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis. 2009;68(suppl 2):ii1-44.
Spondyloarthritis:
Peripheral
ASAS classification criteria for peripheral spondyloarthritis
Arthritis or Enthesitis or Dactylitis
Plus 1 of:
- Psoriasis
- Inflammatory bowel disease
- Preceding infection
- HLA-B27
- Uveitis
- Sacroiliitis on imaging (radiographs or MRI)
(or)
Plus _2 of the remaining:
- Arthritis
- Enthesitis
- Dactylitis
- IBP in the past
- Positive family history for SpA
The criteria are applicable to patients with peripheral arthritis (usually predominantly of the lower limbs or asymmetric arthritis), enthesitis, or dactylitis.
Reference: Rudwaleit M, van der Heijde D, Landewé R, et al. The Assessment of SpondyloArthritis International Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis. 2011;70(1):25-31.
Definition of Spondyloarthritis Features for Use of Assessment of SpondyloArthritis International Society Classification Criteria for Peripheral Spondyloarthritis
Entry Criteria
Arthritis - Current peripheral arthritis compatible with SpA (usually asymmetric or predominant involvement of the lower limb)*
Enthesitis - Current enthesitis*
Dactylitis - Current dactylitis*
* diagnosed clinically by a doctor
Additional SpA Features
- 1. Inflammatory Back Pain (IBP) in the past - IBP in the past according to the rheumatologist’s judgement
- 2. Arthritis - Past or present peripheral arthritis compatible with SpA (usually asymmetric or predominant involvement of the lower limb) diagnosed clinically by a doctor
- 3. Enthesitis - past or present spontaneous pain or tenderness at examination of an enthesis
- 4. Uveitis - Past or present uveitis (anterior) confirmed by an ophthalmologist
- 5. Dactylitis - Past or present dactylitis diagnosed by a doctor
- 6. Psoriasis - Past or present psoriasis diagnosed by a doctor
- 7. IBD - Past or present CD or UC diagnosed by a doctor
- 8. Preceding infection - Urethritis, cervicitis, or diarrhoea within 1 month before the onset of arthritis, enthesitis, or dactylitis
- 9. Family history of SpA - Presence in first- (mother, father, sisters, brothers, children) or second-degree (maternal and paternal grandparents, aunts, uncles, nieces, nephews) relatives of any of the following: (1) ankylosing spondylitis, (2) psoriasis, (3) acute uveitis, (4) reactive arthritis, (5) IBD
- 10. HLA-B27 - Positive testing according to standard laboratory techniques
- 11. Sacroiliitis by imaging - Bilateral grade 2–4 or unilateral grade 3–4 sacroiliitis on plain radiographs, according to the modified New York criteria, or active sacroiliitis on MRI according to the ASAS consensus definition
Reference: Rudwaleit M, van der Heijde D, Landewé R, et al. The Assessment of SpondyloArthritis International Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis. 2011;70(1):25-31.
Reactive arthritis
Diagnostic criteria for reactive arthritis
| Criteria | ||
|---|---|---|
| MAJOR | 1.Arthritis with two of three | a) Asymmetric b) Mono- or oligoarthritis c) Lower limb involvement |
| MAJOR | 2. Preceding symptomatic infection with ³ 1 | a) Enteritis - diarrhoea for at least 1 day, 3 days to 6 weeks before the onset of arthritis b) Urethritis (dysuria or discharge for at least 1 day, 3 days to 6 weeks before the onset of arthritis) |
| Minor | 1.Evidence of triggering infection | a)Positive urine ligase reaction or urethral or cervical swab for Chlamydia trachomatis b) Positive stool culture for enteric pathogens associated with reactive arthritis |
| Minor | 2. Evidence of persistent synovial infection | Positive immunohistology or PCR for Chlamydia |
Definite – 2 Major + ³ 1 minor
Probable – 2 Major or ³ 1 Major + ³ 1 Minor
The identification of the trigger infection is also required.
Reference: Selmi C, Gershwin ME. Diagnosis and classification of reactive arthritis. Autoimmun Rev. 2014;13(4-5):546-549.
Psoriatic arthritis
The CASPAR criteria
To meet the CASPAR (ClASsification criteria for Psoriatic AR thritis)criteria, a patient must have inflammatory articular disease(joint, spine, or entheseal) with ≥3 points from the following five categories:
| Features | Points |
|---|---|
| 1. Psoriasis (PsO) Current history1 Personal history2 Family history PsO3 |
2 1 1 |
| 2. Current Psoriatic nail dystrophy including onycholysis, pitting, and hyperkeratosis | 1 |
| 3. A negative test result for the presence of RF by any method except latex but preferably by ELISA or nephelometry, according to the local laboratory reference range | 1 |
| 4. Either current dactylitis, defined as swelling of an entire digit, or a history of dactylitis recorded by a rheumatologist | 1 |
| 5. Radiographic evidence of juxta-articular new bone formation, appearing as ill-defined ossification near joint margins (but excluding osteophyte formation) on plain radiographs of the hand or foot | 1 |
1 Current psoriasis is defined as psoriatic skin or scalp disease present today as judged by a rheumatologist or dermatologist. 1
2 A personal history of psoriasis is defined as a history of psoriasis that may be obtained from a patient, family physician, dermatologist, rheumatologist, or other qualified health care provider.
A family history of psoriasis is defined as a history of psoriasis in a first- or second-degree relative according to patient report.
Reference: Taylor W, Gladman D, Helliwell P, et al; CASPAR Study Group. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54(8):2665-2673.
Moll and wright Criteria
Presence of PsO and an inflammatory arthritis (peripheral arthritis and/or sacroiliitis or spondylitis) with the absence of serologic tests for Rheumatoid factor
Moll JM, Wright V. Psoriatic arthritis. Semin ArthritisRheum. 1973;3:55-78.
4. SYSTEMIC LUPUS ERYTHEMATOSUS
SYSTEMIC LUPUS INTERNATIONAL COLLABORATING CLINICS (SLICC) CLASSIFICATION CRITERIA FOR SYSTEMIC LUPUS ERYTHEMATOSUS1
The patient must satisfy at least four criteria, including at least one clinical criterion and one immunologic criterion or the patient must have biopsy-proven lupus nephritis in the presence of antinuclear antibodies or anti–double-stranded DNA antibodies.
Clinical criteria
2. Acute cutaneous lupus, including
- Lupus malar rash (do not count if malar discoid)
- Bullous lupus
- Toxic epidermal necrolysis variant of SLE
- Maculopapular lupus rash
- Photosensitive lupus rash
- In the absence of dermatomyositis
Or subacute cutaneous lupus (nonindurated psoriasiform or annular polycyclic lesions that resolve without scarring, although occasionally with post-inflammatory dyspigmentation or telangiectasias)
2. Chronic cutaneous lupus, including
- Classical discoid rash
- Localized (above the neck)
- Generalized (above and below the neck)
- Hypertrophic (verrucous) lupus
- Lupus panniculitis (profundus)
- Mucosal lupus
- Lupus erythematosus tumidus
- Chilblains lupus
- Discoid lupus or lichen planus overlap
3. Oral ulcers
- Palate
- Buccal
- Tongue
Or nasal ulcers
In the absence of other causes, such as vasculitis, Behçet disease, infection (herpes), IBD, reactive arthritis, and acidic foods
4. Nonscarring alopecia (diffuse thinning or hair fragility with visible broken hairs)
In the absence of other causes such as alopecia areata, drugs, iron deficiency, and androgenic alopecia
5. Synovitis involving two or more joints, characterized by swelling or effusion
or tenderness in two or more joints and 30 minutes or more of morning stiffness
6. Serositis
Or pericardial rub
Or pericarditis by ECG
In the absence of other causes, such as infection, uremia, and Dressler pericarditis
- Typical pleurisy for more than 1 day
- Or pleural effusions
- Or pleural rub
- Typical pericardial pain (pain with recumbency improved by sitting forward) for more than 1 day
- Or pericardial effusion
7. Renal
Urine protein-to-creatinine ratio (or 24-hr urine protein) representing 500 mg of protein/24 hr
Or RBC casts
8. Neurologic
- Seizures
- Psychosis
- Mononeuritis multiplex
- In the absence of other known causes such as primary vasculitis
- Myelitis
- Peripheral or cranial neuropathy
- In the absence of other known causes such as primary vasculitis, infection, and diabetes mellitus
- Acute confusional state
- In the absence of other causes, including toxic-metabolic, uremia, drugs
9. Hemolytic anemia
10. Leukopenia (<4000/mm3 at least once)
In the absence of other known causes such as Felty syndrome, drugs, and portal hypertension
Or
Lymphopenia (<1000/mm3 at least once)
In the absence of other known causes such as corticosteroids, drugs, and infection
11. Thrombocytopenia (<100,000/mm3) at least once
In the absence of other known causes such as drugs, portal hypertension, and TTP
Immunologic criteria
- ANA above laboratory reference range
- Anti-dsDNA above laboratory reference range (or >two-fold the reference range if tested by ELISA)
- Anti-Sm: presence of antibody to Sm nuclear antigen
- Antiphospholipid antibody positivity as determined by any of the following:
Positive test result for lupus anticoagulant
False-positive test result for rapid plasma reagin
Medium- or high-titer anticardiolipin antibody level (IgA, IgG, or IgM)
Positive test result for anti–β2-glycoprotein I (IgA, IgG, or IgM)
5. Low complement
Low C3
Low C4
Low CH50
6. Direct Coombs test in the absence of hemolytic anemia
1- Criteria are cumulative and need not be present concurrently.
Reference: Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686.
1997 Update of the 1982 American College of Rheumatology Revised Criteria for the Classification of Systemic Lupus Erythematosus
1. Malar rash
Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds
2. Discoid rash
Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions
3. Photosensitivity
Skin rash as a result of unusual reaction to sunlight by patient history or physician observation
4. Oral ulcers
Oral or nasopharyngeal ulceration, usually painless, observed by physician
5. Nonerosive arthritis
Involving two or more peripheral joints, characterized by tenderness, swelling, or effusion
6. Pleuritis or pericarditis
- A. Pleuritis: convincing history of pleuritic pain or rubbing heard by a physician or evidence of pleural effusion (or)
- B. Pericarditis: documented by ECG or rub or evidence of pericardial effusion
7. Renal disorder
- A. Persistent proteinuria >0.5 g/day or >3+ if quantitation not performed (Or)
- B. Cellular casts: may be RBC, hemoglobin, granular, tubular, or mixed
8. Neurologic disorder
- A. Seizures: in the absence of offending drugs or known metabolic derangements (e.g., uremia, ketoacidosis, electrolyte imbalance) (Or)
- B. Psychosis: in the absence of offending drugs or known metabolic derangements (e.g., uremia, ketoacidosis, electrolyte imbalance)
9. Hematologic disorder
- A. Hemolytic anemia with reticulocytosis (Or)
- B. Leukopenia: <4000/mm3 total on two or more occasions (Or)
- C. Lymphopenia: <1500/mm3 on two or more occasions (Or)
- D. Thrombocytopenia: <100,000/mm3 in the absence of offending drugs
10. Immunologic disorder
- A. Anti-DNA: antibody to native DNA in abnormal titer (Or)
- B. nti-Sm: presence of antibody to Sm nuclear antigen (Or)
- C. Positive finding of antiphospholipid antibodies on:
- 1. An abnormal serum level of IgG or IgM anticardiolipin antibodies
- 2. A positive test result for lupus anticoagulant using a standard method (Or)
- 3. A false-positive test result for at least 6 months confirmed by Treponema pallidum immobilization or fluorescent treponemal antibody absorption test
11. Positive antinuclear antibody
An abnormal titer of antinuclear antibody by immunofluorescence or an equivalent assay at any point in time and in the absence of drugs
For the purpose of identifying patients in clinical studies, a person is said to have systemic lupus erythematosus if any 4 or more of the 11 criteria are present, serially or simultaneously, during any interval of observation.
Reference: Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus [letter]. Arthritis Rheum. 1997;40:1725; and Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271.
THE ACR/ EULAR CRITERIA FOR CLASSIFICATION OF SLE, 2018
Everyone must have ANA (antinuclear antibody in blood test) with titer of at least 80.
Total scoreof at least 10 Required for SLE, please calculate below:
- 1. For each criterion, do not score if a more likely cause for the symptom exists.
- 2. Occurrence of a criterion on at least one occasion is sufficient.
- 3. Criteria need not occur simultaneously.
- 4. At least one clinical criterion must be present.
- 5. Within each domain, only the highest weighted criterion is counted toward the total score.
| Clinical domains and criteria | Weight | Immunologic domains and criteria | Weight |
|---|---|---|---|
| Constitutional domain | Antiphospholipid antibody domain | ||
| Fever >38.3 ⁰C | 2 | Anti-cardiolipin IgG >40 GPL units or Anti- β2GP-I IgG >40 units or lupus anticoagulant positive | 2 |
| Cutaneous domain | Complement proteins domain | ||
| Fever >38.3 ⁰C | 2 | Anti-cardiolipin IgG >40 GPL units or Anti- β2GP-I IgG >40 units or lupus anticoagulant positive | 2 |
| Non-scarring alopecia Oral ulcers Subacute cutaneous or Discoid lupus Acute cutaneous lupus |
2 2 4 6 |
Low C3 or Low C4 Low C3 and Low C4 at the same time |
3 4 |
| Arthritis domain | Highly specific antibodies domain | ||
| Synovitis in ≥2 joints or tenderness in ≥2 joints and ≥30 minutes of morning stiffness | 6 | Anti dsDNA antibody Anti Smith antibody |
6 6 |
| Neurologic domain | Hematologic domain | ||
| Delirium Psychosis Seizure |
2 3 5 |
Leucopenia (<4000/ cu.mm) Thrombocytopenia Autoimmune hemolysis |
3 4 4 |
| Serositis domain | |||
| Pleural or pericardial effusion Acute pericarditis |
5 6 |
||
| Renal domain | |||
| Proteinuria >0.5g/ 24h Renal biopsy with class II or V nephritis Renal biopsy with class III or IV nephritis |
4 8 10 |
||
*Classify as SLE if total score ≥10 points
5. Antiphospholipid Antibody Syndrome
Revised classification criteria for antiphospholipid syndrome
Antiphospholipid antibody syndrome (APS) is present if at least one of the clinical criteria and one of the laboratory criteria that follow are met.*
Clinical criteria
1. Vascular thrombosis#
ne or more clinical episodes‡of arterial, venous, or small vessel thrombosis§in any tissue or organ. Thrombosis must be confirmed by objective validated criteria (i.e., unequivocal findings of appropriate imaging studies or histopathology). For histopathologic confirmation, thrombosis should be present without significant evidence of inflammation in the vessel wall
2. Pregnancy morbidity
a) One or more unexplained deaths of a morphologically normal foetusat or beyond the 10th week of gestation with normal foetal morphology documented by ultrasound or by direct examination of the foetusOR
b) One or more premature births of a morphologically normal neonate before the 34th week of gestation because of (i) eclampsia or severe preeclampsia defined according to standard definitions1 or (ii) recognized features of placental insufficiency∥or
c) Three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes excluded.
In studies of populations of patients who have more than one type of pregnancy morbidity, investigators are strongly encouraged to stratify groups of subjects according to a, b, or c above.
Laboratory criteria
| Reference Range | |
|---|---|
| 1) LA in plasma on two or more occasions at least 12 weeks apart, detected according to the guidelines of the International Society on Thrombosis and Haemostasis (Scientific Subcommittee on LAs/phospholipid-dependent antibodies)2,3. | Positive |
| 2. aCL antibody of IgG and/or IgM isotype in serum or plasmapresent in medium or high titer on two or more occasions, at least 12 weeks apart, measured by a standardized ELISA4-6 | >40 GPL or MPL, or > the 99th percentile |
| 3. Anti–B2 glycoprotein-I antibody of IgG and/or IgM isotype in serum or plasmapresent on two or more occasions, at least 12 weeks apart, measured by a standardized ELISA, according to recommended procedures7 | > the 99th percentile |
*Classification of APS should be avoided if less than 12 weeks or more than 5 years separates the positive aPL test and the clinical manifestation.
#Coexisting inherited or acquired factors for thrombosis are not reasons for excluding patients from APS trials. However, two subgroups of APS patients should be recognized, according to (a) the presence and (b) the absence of additional risk factors for thrombosis. Indicative (but not exhaustive) such cases include age (>55 years in men and >65 years in women) and the presence of any of the established risk factors for cardiovascular disease (hypertension, diabetes mellitus, elevated LDL or low HDL cholesterol, cigarette smoking, family history of premature cardiovascular disease, BMI ≥30 kg/m2, microalbuminuria, estimated GFR <60 mL min−1, inherited thrombophilia, oral contraceptives, nephrotic syndrome, malignancy, immobilization, and surgery. Thus, patients who fulfil criteria should be stratified according to contributing causes of thrombosis.
‡A thrombotic episode in the past could be considered as a clinical criterion, provided that thrombosis is proved by appropriate diagnostic means and that no alternative diagnosis or cause of thrombosis is found.
Superficial venous thrombosis is not included in the clinical criteria.
∥Generally accepted features of placental insufficiency include (i) abnormal or nonreassuring foetal surveillance test(s) (e.g., a nonreactive nonstress test, suggestive of foetal hypoxemia), (ii) abnormal Doppler flow velocimetry waveform analysis suggestive of foetal hypoxemia (e.g., absent end-diastolic flow in the umbilical artery), (iii) oligohydramnios (e.g., an amniotic fluid index of ≤5 cm), or (iv) a postnatal birth weight less than the 10th percentile for gestational age.
¶ Investigators are strongly advised to classify patients with APS in studies into one of the following categories: I, more than one laboratory criterion present (any combination); IIa, LA present alone; IIb, aCL antibody present alone; and IIc, anti-β2 glycoprotein-I antibody present alone.
- 1. American College of Obstetricians and Gynecologists. Diagnosis and Management of Preeclampsia and Eclampsia. ACOG Practice Bulletin No. 33. Washington, DC: American College of Obstetricians and Gynecologists; 2002
- 2. Wisloff F, Jacobsen EM, Liestol S. Laboratory diagnosis of the antiphospholipid syndrome. Thromb Res. 2002;108:263-271.
- 3. Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost. 1995;74:1185-1190.
- 4. Tincani A, Allegri F, Sanmarco M, et al. Anticardiolipin antibody assay: a methodological analysis for a better consensus in routine determinations—a cooperative project of the European Antiphospholipid Forum. Thromb Haemost. 2001;86:575-583.
- 5. Harris EN, Pierangeli SS. Revisiting the anticardiolipin test and its standardization. Lupus. 2002;11:269-275.
- 6. Wong RC, Gillis D, Adelstein S, et al. Consensus guidelines on anti-cardiolipin antibody testing and reporting. Pathology. 2004;36:63-68.
- 7. Reber G, Tincani A, Sanmarco M, et al. Proposals for the measurement of anti-beta2-glycoprotein I antibodies. Standardization group of the European Forum on Antiphospholipid Antibodies. J Thromb Haemost. 2004;2:1860-1862.
Reference: Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
Preliminary Criteria for the Classification of Catastrophic Antiphospholipid Syndrome (APS)
- 1. Evidence of involvement of three or more organs, systems, or tissues*
- 2. Development of manifestations simultaneously or in less than 1 week
- 3. Confirmation by histopathology of small vessel occlusion in at least one organ or tissue†
- 4. Laboratory confirmation of the presence of antiphospholipid antibody (lupus anticoagulant or anticardiolipin or anti–β2-glycoprotein I antibodies)‡
Definite Catastrophic APS- All four criteria
Probable Catastrophic APS
Criteria 2 through 4 and two organs, systems, or tissues involved
Criteria 1 through 3, except no confirmation 6 week apart owing to early death of patient not tested before catastrophic episode Criteria 1, 2, and 4
Criteria 1, 3, and 4 and development of a third event more than 1 week but less than 1 month after the first, despite anticoagulation
*Usually, clinical evidence of vessel occlusions, confirmed by imaging techniques when appropriate. Renal involvement is defined by a 50% rise in serum creatinine, severe systemic hypertension, proteinuria, or some combination of these.
†For histopathologic confirmation, significant evidence of thrombosis must be present, although vasculitis may coexist occasionally.
‡If the patient has not been diagnosed previously with APS, laboratory confirmation requires that the presence of antiphospholipid antibody be detected on two or more occasions at least 6 weeks apart (not necessarily at the time of the event), according to proposed preliminary criteria for the classification of APS.
Reference: Asherson RA, Cervera R, de Groot PG, et al: Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines, Lupus 12:530–534, 2003.
6. Systemic Sclerosis
Establishing a Diagnosis of Scleroderma
1980 ACR Criteria
Must have (a) or two of (b), (c), or (d):
- 1. Proximal SSc (proximal to MCPs/MTPs)
- 2. gital pits
- 3. Sclerodactyly
- 4. Pulmonary fibrosis (chest radiograph; HRCT)
CREST Criteria
Must have three of the five features:
- 1. Calcinosis
- 2. Raynaud’s phenomenon
- 3. Esophageal dysmotility
- 4. Sclerodactyly
- 5. Telangiectasias
Minor Criteria*†
Must have all three:
- 1. Definite Raynaud’s phenomenon
- 2. Abnormal capillary loops
- 3. Specific scleroderma autoantibody
*Some experts continue to classify patients with minor criteria only as
undifferentiated connective disease with scleroderma features.
†Autoantibody includesanticentromere,
Anti-topoisomerase 1 (Scl-70), and anti-RNA polymerase III.
2013 American College of Rheumatology/European League Against Rheumatism Criteria for the Classification of Systemic Sclerosis*
Item Sub-item Weight or score† Skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints (sufficient criterion) 9 Skin thickening of the fingers (only count the higher score) Puffy fingers
Sclerodactyly of the fingers (distal to the metacarpophalangeal joints but proximal to the proximal interphalangeal joints)2
4Fingertip lesions (only count the higher score) Digital tip ulcers
Fingertip pitting scars2 br 3 Telangiectasia 2 Abnormal nailfold capillaries 2 Raynaud phenomenon 3 Pulmonary arterial hypertension or interstitial lung disease (maximum score is 2) Pulmonary arterial hypertension
Interstitial lung disease2
2SSc-related autoantibodies (anticentromere, anti– topoisomerase I [anti–Scl-70], anti–RNA polymerase III) (maximum score is 3) Anticentromere
Anti–topoisomerase I
Anti–RNA polymerase III3 *These criteria are applicable to any patient considered for inclusion in a systemic sclerosis (SSc) study. The criteria are not applicable to patients with skin thickening sparing the fingers or to patients who have a scleroderma-like disorder that better explains their manifestations (e.g., nephrogenic sclerosing fibrosis, generalized morphea, eosinophilic fasciitis, scleredema diabeticorum, scleromyxedema, erythromyalgia, porphyria, lichen sclerosis, graft-versus-host disease, diabetic cheiroarthropathy).
†The total score is determined by adding the maximum weight (score) in each category. Patients with a total score of ≥9 are classified as having definite SSc.
Reference: van den Hoogen F, Khanna D, Fransen J, Johnson SR, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. Nov;65(11):2737-2747.
Definitions of Items and Subitems in the American College of Rheumatology/European League Against Rheumatism Criteria for the Classification of Systemic Sclerosis
Item Definition Skin thickening Skin thickening or hardening not caused by scarring after injury, trauma, and so on Puffy fingers Swollen digits: a diffuse, usually nonpitting increase in soft tissue mass of the digits extending beyond the normal confines of the joint capsule. Normal digits are tapered distally with the tissues following the contours of the digital bone and joint structures. Swelling of the digits obliterates these contours. Not from other causes such as inflammatory dactylitis. Fingertip ulcers or pitting scars Ulcers or scars distal to or at the proximal interphalangeal joint not thought to be caused by trauma. Digital pitting scars are depressed areas at digital tips as a result of ischemia rather than trauma or exogenous causes. Telangiectasia Telangiectasias are visible macular dilated superficial blood vessels that collapse upon pressure and fill slowly when pressure is released. Telangiectasias in a scleroderma-like pattern are round and well demarcated and found on the hands, lips, or inside of the mouth or are large mat like telangiectasias. Distinguishable from rapidly filling spider angiomas with central arteriole and from dilated superficial vessels. Abnormal nailfold capillary pattern consistent with systemic sclerosis Enlarged capillaries or capillary loss with or without pericapillary hemorrhages at the nailfold; may also be seen on the cuticle Pulmonary arterial hypertension Pulmonary arterial hypertension diagnosed by right-sided heart catheterization according to standard definitions Interstitial lung disease Pulmonary fibrosis seen on high-resolution computed tomography or chest radiography, most pronounced in the basilar portions of the lungs, or occurrence of “Velcro” crackles on auscultation not from another cause such as congestive heart failure Raynaud phenomenon Self-reported or reported by a physician, with at least a two-phase color change in finger(s) and often toe(s) consisting of pallor, cyanosis, or reactive hyperemia in response to cold exposure or emotion; usually one phase is pallor Systemic sclerosis– related autoantibodies Anticentromere antibody or centromere pattern seen on antinuclear antibody testing, antitopoisomerase I antibody (also known as anti-Scl-70 antibody), or anti-RNA polymerase III antibody; positive according to local laboratory standards Reference: van den Hoogen F, Khanna D, Fransen J, Johnson SR, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013 Nov65(11):2737-2747.
7. IDIOPATHIC INFLAMMATORY MYOSITIS
BOHAN AND PETER CRITERIA FOR THE DIAGNOSIS OF POLYMYOSITIS AND DERMATOMYOSITIS
Full criteria can be found in Bohan A, Peter JB. Polymyositis and dermatomyositis (parts 1 and 2). N Engl J Med. 1975;292:34
Definite PM defined as all first four elements, probable PM as three of first four and possible PM as two of first four; definite DM defined as rash plus three other elements, probable DM as rash plus two others and possible DM as rash plus one other. DM, Dermatomyositis; PM, Polymyositis.
Reference: Lunberg IE, Miller FW, et al. Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med. 2016; 280:39-51.
The European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) classification criteria for adult and juvenile idiopathic inflammatory myopathies (IIMs)
When no better explanation for the symptoms and signs exists, these classification criteria can be used
Score points Variable Without muscle biopsy With muscle biopsy Definition Age of onset
1. Age of onset of first symptom assumed to be related to the disease ≥18 years and <40 years
2. Age of onset of first symptom assumed to be related to the disease ≥40 years1.3
2.11.5
2.218 ≤ age (years) at onset of first symptom assumed to be related to the disease <40
Age (years) at onset of first symptom assumed to be related to the disease ≥40Muscle weakness
1. Objective symmetric weakness, usually progressive, of the proximal upper extremities
2. Objective symmetric weakness, usually progressive, of the proximal lower extremities
3. Neck flexors are relatively weaker than neck extensors
4. In the legs, proximal muscles are relatively weaker than distal muscles0.7
0.8
1.9
0.90.7
0.5
0.5
1.2Weakness of proximal upper extremities as defined by manual muscle testing or other objective strength testing, which is present on both sides and is usually progressive over time
Weakness of proximal lower extremities as defined by manual muscle testing or other objective strength testing, which is present on both sides and is usually progressive over time
Muscle grades for neck flexors are relatively lower than neck extensors as defined by manual muscle testing or other objective strength testing
Muscle grades for proximal muscles in the legs are relatively lower than distal muscles in the legs as defined by manual muscle testing or other objective strength testingSkin manifestations
1. Heliotrope rash
2. Gottron’s papule
3. Gottron’s rash3.1
2.1
3.33.2
2.7
3.7Purple, lilac-colored, or erythematous patches over the eyelids or in a periorbital distribution, often associated with periorbital edema
Erythematous to violaceous papules over the extensor surfaces of joints, which are sometimes scaly. May occur over the finger joints, elbows, knees, malleoli, and toes
Erythematous to violaceous macules over the extensor surfaces of joints, which are not palpableOther clinical manifestations
Dysphagia or esophageal dysmotility0.7 0.6 Difficulty in swallowing or objective evidence of abnormal motility of the esophagus Laboratory measurements
1. Anti–Jo-1 (anti–histidyl–transfer RNA synthetase) autoantibody present
2. Elevated serum levels of creatine kinase (CK) or lactate dehydrogenase (LDH) or aspartate aminotransferase (ASAT/AST/SGOT) or alanine aminotransferase (ALAT/ALT/SGPT) above upper limits of normal3.9
1.33.8
1.4Autoantibody testing in serum performed with standardized and validated test, showing positive result
The most abnormal test values during the disease course (highest absolute level of enzyme) above the relevant upper limit of normalMuscle biopsy features-presence of:
1. Endomysial infiltration of mononuclear cells surrounding, but not invading myofibers
2. Perimysial and/or perivascular infiltration of mononuclear cells
3. Perifascicular atrophy
4. Rimmed vacuoles1.7
1.7
1.9
3.1Muscle biopsy reveals endomysial mononuclear cells abutting the sarcolemma of otherwise healthy, non-necrotic muscle fibers, but there is no clear invasion of the muscle fibers
Mononuclear cells are located in the perimysium and/or located around blood vessels (in either perimysial or endomysial vessels)
Muscle biopsy reveals several rows of muscle fibers, which are smaller in the perifascicular region than fibers more centrally located
Rimmed vacuoles are bluish by haematoxylin and eosin staining and reddish by modified Gomori trichrome stainProbability of IIM without muscle biopsy = 1/ [1+ exponential(5.33-score)]
Probability of IIM including muscle biopsy = 1/ [1+ exponential(6.49-score)]
Classification as IIM Score without biopsy Score with biopsy Not classified < 5.3 <6.5 Possible 5.3 – 5.49 6.5 - 6.69 Probable 5.5 – 7.49 6.7 – 8.69 Definite ³ 7.5 ³ 8.7 Reference: Lundberg IE, Tjärnlund A, Bottai M, et al. EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. ARTHRITIS & RHEUMATOLOGY. Vol. 69, No. 12, December 2017, pp 2271–2282.
“GRIGGS CRITERIA” FOR INCLUSION BODY MYOSITIS
1. Characteristic features
A. Clinical
Proximal and distal of arms and legs and must exhibit at least one of:
D. Laboratory features
1. CK <12× normal
2. Muscle biopsy
Diagnostic criteria: (i) definite inclusion body myositis (IBM) = all muscle biopsy features. None of the clinical or laboratory features are mandatory; (ii) possible IBM = partial invasion without other pathologic features + characteristic clinical and other laboratory features.
Reference: Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995;38:705-713; and Hilton-Jones D, Brady S. Diagnostic criteria for inclusion body myositis. J Intern Med. 2016;280(1):52-62.
The European Neuromuscular Centre Inclusion Body Myositis (IBM) 2011 Research Diagnostic Criteria
Clinical and laboratory features Classification Pathologic features Duration >12 months
Age at onset >45 yearsClinicopathologically defined IBM All of the following:
Endomysial inflammatory infiltrate
Rimmed vacuoles Protein accumulation* or 15- to 18-nm filamentsKnee extension weakness ≥ hip flexion weakness
and/or
Finger flexion weakness > shoulder abduction weakness and
Serum CK no greater than 15 × ULNDuration >12 months
Age at onset >45 yearsClinically defined IBM One or more, but not all, of: Endomysial inflammatoryinfiltrate
Upregulation of MHC class I Rimmed vacuolesKnee extension weakness ≥ hip flexion weakness
and
Finger flexion weakness > shoulder abduction weakness sCK no greater than 15*ULNProtein accumulation* or 15- to 18-nm filaments Duration >12 months
Age at onset >45 yearsProbable IBM One or more, but not all, of: Endomysial inflammatory infiltrate
Upregulation of MHC class I Rimmed vacuolesKnee extension weakness ≥ hip flexion weakness
or
Finger flexion weakness > shoulder abduction weakness sCK no greater than 15*ULNProtein accumulation* or 15- to 18-nm filaments MHC, Major Histocompatibility Complex; sCK, serum creatine kinase; ULN, Upper limit of normal
*Demonstration of amyloid or other protein accumulation by established methods (e.g., for amyloid Congo red, crystal violet, thioflavin T/S or for other proteins p62, SMI-31, TDP-43). Current evidence favours p62 in terms of sensitivity and specificity but the literature is limited, and further work is required.
Reference: Rose MR, et al; ENMC IBM Working Group.188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044-1055.
8. MIXED CONNECTIVE TISSUE DISEASE
Alarcòn Segovia Criteria
Serologic criteria Anti-RNP at hemagglutination titer of ≥1 : 1600 Clinical criteria 1. Swollen hands
2. Synovitis
3. Myositis (biologically proven)
4. Raynaud’s phenomenon
5. AcrosclerosisMCTD present if: Serologic criterion accompanied by 3 or more clinical criteria, one of which must include synovitis or myositis Kahn Criteria
Serologic criteria High-titer anti-RNP corresponding to a speckled ANA of ≥1 : 1200 titer Clinical criteria 1. Swollen fingers
2. Synovitis
3. Myositis
4. Raynaud’s phenomenonMCTD present if Serologic criterion accompanied by Raynaud’s phenomenon and 2 or more of the 3 remaining clinical criteria Reference: Alarcon-Segovia D, Cardiel MH: Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients, J Rheumatol 16(3):328–334, 1989.
For Kasukawa and Sharp’s criteria refer to review article on MCTD by Yolanda Farhey and Evelyn V. Hess at onlinelibrary.wiley.com/doi/pdf/10.1002/art.1790100508
9. GIANT CELL ARTERITIS
1990 American College of Rheumatology Criteria for the Classification of Giant Cell (Temporal) Arteritis*
*For purposes of classification, a patient shall be said to have giant cell (temporal) arteritis if at least three of these five criteria are present.
Reference: Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122-1128.
Polymyalgia rheumatica
Required criteria: age ≥50 years, bilateral shoulder aching, and abnormal CRP or ESR*
Points without US (0-6) Points with US (0-8) Morning stiffness duration ≥45 minutes 2 2 Hip pain or limited range of motion 1 1 Absence of RF or ACPA 2 2 Absence of other joint involvement 1 1 At least one shoulder with subdeltoid bursitis or biceps tenosynovitis or glenohumeral synovitis (either posterior or axillary) and at least one hip with synovitis or trochanteric bursitis NA 1 Both shoulders with subdeltoid bursitis, biceps tenosynovitis, or glenohumeral synovitis NA 1 *A score of 4 or more is categorized as polymyalgia rheumatica (PMR) in the algorithm without ultrasound (US), and a score of 5 or more is categorized as PMR in the algorithm with US.
Reference: Dasgupta B, Cimmino MA, Kremers HM, et al. 2012 Provisional classification criteria for polymyalgia rheumatica: a European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis Rheum. 2012;64(4):943-954.
10. TAKAYASU ARTERITIS
1990 American College of Rheumatology Criteria for the Classification of Takayasu Arteritis
Criterion Definition Age at disease onset ≤40 years Development of symptoms or findings related to Takayasu arteritis at age ≤40 years Claudication of extremities Development and worsening of fatigue and discomfort in muscles of one or more extremity while in use, especially the upper extremities Decreased brachial artery pulse Decreased pulsation of one or both brachial arteries BP difference >10 mm Hg Difference of >10 mm Hg in systolic BP between arms Bruit over subclavian arteries or aorta Bruit audible on auscultation over one or both subclavian arteries or abdominal aorta Arteriogram abnormality Arteriographic narrowing or occlusion of the entire aorta, its primary branches, or large arteries in the proximal upper or lower extremities, not caused by arteriosclerosis, fibromuscular dysplasia, or similar causes; changes usually focal or segmental Patient shall be said to have Takayasu arteritis if at least three of these six criteria are present. The presence of any three or more criteria yields a sensitivity of 90.5% and a specificity of 97.8%.
Reference: Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33(8):1129-1134.
Ishikawa criteria [ 2 Major or One Major with ≥2 minor or ≥4 minor(representing vessel involvement)
Obligatory Age of onset ≤ 40 years and symptoms duration > 1 month Major Involvement of Left mid-SCA Involvement of Right mid-SCA Y/ N Minor Raised ESR Carotid artery tenderness Hypertension AR or annuloaortic ectasia Pulmonary artery lesions Left mid-common carotid lesion Descending thoracic aorta lesion Abdominal aorta lesion AR, aortic regurgitation; ESR, Erythrocyte sedimentation rate; SCA, sub-clavian artery
11. KAWASAKI DISEASE
Classic Kawasaki disease (KD) is diagnosed in the presence of fever for at least 5 days (the day of fever onset is taken to be the first day of fever) together with at least four of the five following principal clinical features. In the presence of ≥4 principal clinical features, particularly when redness and swelling of the hands and feet are present, the diagnosis of KD can be made with 4 days of fever, although experienced clinicians who have treated many patients with KD may establish the diagnosis with 3 days of fever in rare cases:
A careful history may reveal that ≥1 principal clinical feature was present during the illness but resolved by the time of presentation.
Patients who lack full clinical features of classic KD are often evaluated for incomplete KD. If coronary artery abnormalities are detected, the diagnosis of KD is considered confirmed in most cases.
Laboratory tests typically reveal normal or elevated white blood cell count with neutrophil predominance and elevated acute phase reactants such as C-reactive protein and erythrocyte sedimentation rate during the acute phase. Low serum sodium and albumin levels, elevated serum liver enzymes, and sterile pyuria can be present. In the second week after fever onset, thrombocytosis is common.
Reference: McCrindle BW, Rowley AH, Newburger JW, et al; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Surgery and Anesthesia; and Council on Epidemiology and Prevention. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals from the American Heart Association. Circulation. 2017;135(17):e927-e999.
EUROPEAN LEAGUE AGAINST RHEUMATISM/PAEDIATRIC RHEUMATOLOGY EUROPEAN SOCIETY CLASSIFICATION CRITERIA FOR KAWASAKI DISEASE
Fever persisting for at least 5 days (mandatory criterion) plus four of the following five features:
In the presence of coronary artery involvement (detected on echocardiography) and fever, fewer than four of the remaining five criteria are sufficient (the exact number of criteria required is to be defined in the validation phase).
Reference: Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65(7):936-941.
12. HENOCH-SCHÖNLEIN PURPURA and IgA VASCULITIS
European League Against Rheumatism/Pediatric Rheumatology International Trials Organisation/Paediatric Rheumatology European Society Henoch-Schönlein Purpura Criteria (with Glossary) and Classification Definition
Criterion Glossary Purpura (mandatory criterion) Purpura (commonly palpable and in crops) or petechiae, with lower limb predominance* not related to thrombocytopenia 1. Abdominal pain Diffuse abdominal colicky pain with acute onset assessed by history and physical examination; may include intussusception and GI bleeding 2. Histopathology Typically, leukocytoclastic vasculitis with predominant IgA deposit or proliferative glomerulonephritis with predominant IgA deposit 3. Arthritis and arthralgias Arthritis of acute onset defined as joint swelling or joint pain with limitation on motion
Arthralgia of acute onset defined as joint pain without joint swelling or limitation on motion4. Renal involvement Proteinuria >0.3 g/24 hr or >30 mmol/mg of urine albumin/creatinine ratio on a spot morning sample
Hematuria or RBC casts: >5 RBCs per high-power field or red blood cell casts in the urinary sediment or ≥2+ on dipstick1990 American College of Rheumatology Criteria for the Classification of Henoch-Schönlein Purpura
Criterion Definition Palpable purpura Slightly raised “palpable” hemorrhagic skin lesions, not related to thrombocytopenia Age ≤20 years at disease onset Patient 20 years or younger at onset of first symptoms Bowel angina Diffuse abdominal pain, worse after meals, or the diagnosis of bowel ischemia, usually including bloody diarrhoea Wall granulocytes on biopsy Histologic changes showing granulocytes in the walls of arterioles or venules For purposes of classification, a patient shall be said to have Henoch-Schönlein purpura if at least two of these four criteria are present.
Reference: Mills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheum. 1990;33(8):1114-1121.
13. BEHҪET DISEASE
International Criteria for Behçet Disease
Point score system: scoring ≥4 indicates Behçet disease diagnosis
Sign or symptom Points Ocular lesions 2 Genital aphthae 2 Oral aphthae 2 Skin lesions 1 Vascular manifestations 1 Neurologic manifestations 1 *Positive pathergy test 1 *Pathergy test is optional; when it is done, 1 additional point is given for a positive result.
Reference: Davatchi F, Assaad-Khalil S, Calamia KT, et al, for the International Team for the Revision of the International Criteria for Behçet’s Disease (ITR-ICBD). The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338-347.
International Study Group criteria for Behçet Disease
Recurrent oral ulceration Minor aphthous, major aphthous, or herpetiform ulcers observed by the physician or reliably described by the patient, which recurred at least three times in one 12-month period Plus two of Recurrent genital ulceration Aphthous ulceration or scarring observed by the physician or reliably described by the patient Eye lesions Anterior or posterior uveitis or cells in the vitreous body on slit-lamp examination or retinal vasculitis observed by an ophthalmologist Skin lesions Erythema nodosum observed by a physician or patient, pseudofolliculitis, or papulopustular lesions or acneiform nodules observed by a physician in postadolescent patients not on glucocorticoid treatment Positive Pathergy test Read by physician at 24–48 hours Reference: International Study Group for Behcet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet. 1990;335:1078-1080.
Mason and Barnes Criteria for a Diagnosis of Behçet Disease – At least3 Major or 2 Major + 2 Minor
Major
Minor
From R.M. Mason, C.G. Barnes, Behçet’s syndrome with arthritis,Ann. Rheum. Dis. 28 (1969) 95–103
14. CRYOGLOBULINEMIA
CLASSIFICATION CRITERIA FOR CRYOGLOBULINEMIC VASCULITIS
i. Questionnaire item: at least two of the following:
iv. Clinical item: at least three of the following four (present or past)
Constitutional symptoms Fatigue Low-grade fever (37°–37.9°C >10 days; no other cause)
Fever (>38°C; no other cause) FibromyalgiaArticular involvement Arthralgias
ArthritisVascular involvement Purpura
Skin ulcers
Necrotizing vasculitis
Hyperviscosity syndrome
Raynaud phenomenonNeurologic involvement Peripheral neuropathy
Cranial nerve involvement
Vasculitic CNS involvementv. Laboratory item: at least two of the following three (present)†
†The fulfilment of the laboratory item in a patient satisfying the criteria highlights the possible presence of cryoglobulinaemic vasculitis even in the absence of serum cryoglobulins by initial testing.
*Satisfied if at least two of three items (questionnaire, clinical, laboratory) are positive. The patient must be positive for serum cryoglobulins in at least two determinations at a ≥12-week interval.
Reference: De Vita S, Soldano F, Isola M, et al. Preliminary classification criteria for the cryoglobulinaemic vasculitis. Ann Rheum Dis. 2011;70(7):1183-1190; and Quartuccio L, Isola M, Corazza L, et al. Validation of the classification criteria for cryoglobulinaemic vasculitis. Rheumatology (Oxford). 2014;53(12):2209-2213.
15. HYPERSENSITIVITY VASCULITIS
1990 American College of Rheumatology Criteria and Definitions Used for the Classification of Hypersensitivity Vasculitis
Criteria Definition Age at disease onset >16 years Development of symptoms after age 16 years Medication at disease onset Medication was taken at the onset of symptoms that may have been a precipitating factor Palpable purpura Slightly elevated purpuric rash over one or more areas of the skin; does not blanch with pressure and is not related to thrombocytopenia Maculopapular rash Flat and raised lesions of various sizes over one or more areas of the skin Polymorphonuclear neutrophils in vessel wall Biopsy demonstrating granulocytes in the wall of a venule or arteriole Eosinophils in biopsy Biopsy demonstrating eosinophils in a venule or arteriole at any location For purposes of classification, a patient shall be said to have hypersensitivity vasculitis if at least three of these criteria are present.
Reference: Calabrese LH, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of hypersensitivity vasculitis. Arthritis Rheum. 1990;33(8):1108-1113.
16. ANCA Vasculitis
Granulomatosis with polyangiitis
1990 American College of Rheumatology Criteria for the Classification of Granulomatosis with Polyangiitis (Wegener Granulomatosis)
*- a patient shall be said to have Wegener granulomatosis if at least two of these four criteria are present
Reference: Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990;33:1101-1107.
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome)
1990 American College of Rheumatology Criteria for the Classification of Churg-Strauss Syndrome
Four out of Six
Asthma
Eosinophilia >10%
Neuropathy, mono- or poly-
Pulmonary infiltrates
Nonfixed paranasal sinus abnormality
Extravascular eosinophils
Criterion Definition Asthma History of wheezing or diffuse high-pitched rales on expiration Eosinophilia Eosinophilia >10% in WBC differential count Mono or polyneuropathy Development of mononeuropathy, multiple mononeuropathies, or polyneuropathy (i.e., glove-and-stocking distribution) attributable to a systemic vasculitis Pulmonary infiltrates, non-fixed Migratory or transitory pulmonary infiltrates on radiographs (not including fixed infiltrates), attributable to a systemic vasculitis Paranasal sinus abnormality History of acute or chronic paranasal sinus pain or tenderness or radiographic opacification of the paranasal sinuses Extravascular eosinophils Biopsy including artery, arteriole, or venule, showing accumulations of eosinophils in extravascular areas *- a patient shall be said to have Churg-Strauss syndrome if at least four of these six criteria are positive. The presence of any four or more of the six criteria yields a sensitivity of 85% and a specificity of 99.7%.
Reference: Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33:1094-1100.
17. Polyarteritis Nodosa
3 of the 10 ACR criteria should be present when a radiographic or pathological diagnosis of vasculitis is made
Reference
Lightfoot RW Jr, Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. 1990 Aug. 33(8):1088-93
18. MACROPHAGE ACTIVATION SYNDROME
Classification criteria - MAS 2014 [sJIA] * 2016 [sJIA] jSLE [≥ 1 clinical + ≥ 2lab] Laboratory[Lab] criteria
- Platelet <2.62 lac/m
- SGOT > 59 U/L
- WBC <4000/ml
- Fibrinogen ≤ 2.6 g/LA febrile patient with sJIA with ferrtin > 684ng/ml and any two of the following
- Platelet ≤ 1.81 lac/ml
- SGOT > 48 U/L
- TG > 156 mg/dl
- Fibrinogen ≤ 360 mg/dlClincal
- Fever (>380C)
Hepatomegaly [3 cm below the costal margin(BCM)]
. Splenomegaly (3 cm BCM)
. Haemorrhagic manifestations
. Central nervous system dysfunctionClinical criteria
- CNS dysfunction
- Haemorrhages
- Hepatomegaly (≥3 cm below the costal margin)
Histopathological criterion: Evidence of macrophage hemophagocytosis is found in the bone marrow aspirate sample in doubtful cases. *2 Lab or 1 Lab + 1 Clinical criteriaExclude other causes like infectious hepatitis , ITP, famililial hyperlipidemia Lab criteria
Cytopenia affecting 2 or more cell
ineages (WBC ≤ 4000/ml, Hb≤9g/dl or platelet ≤ 1.5lac/ml)
. SGOT (>40 units/l)
. LDH (>567 units/l)
. Fibrinogen ≤ 1.5 gm/l)
. Triglycerides >178 mg/dl)
. Ferritin >500 mg/l)Other important lab parameters – falling ESR, High fibrin degradation products, d-dimer, high PT/APTT, hyponatremia , low albumin, anemia , high LDH, high sIL2αR/sCD25≥ 2400 U/ml, sCD163, low complements in MAS complicating sJIA. Ferritin/ESR ratio > 80 19. ADULT ONSET STILL’S DISEASE
Classification criteria by Yamaguchi et al
Major criteria:
Minor criteria:
Exclusion criteria:
5 criteria: at least 2 major and 3 minor
Reference: Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424e30.
Classification criteria by Fautrel et al
Major criteria:
Minor criteria:
4 major (or) 3 major and 2 minor criteria are required for classification.
Reference: Fautrel B, Zing E, Golmard JL, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002;81:194e200.
20. RELAPSING POLYCHONDRITIS
McAdam et al1
(Requirement: three of six criteria)
Damiani and Levine2
(Requirement: any of these)
Michet et al3
(Requirement: any of these)
References:
From Longo L, Greco A, Rea A, et al. Relapsing polychondritis: a clinical update. Autoimmun Rev. 2016;15(6):539-543
21. IgG4 Related Disease
COMPREHENSIVE CLINICAL DIAGNOSTIC CRITERIA FOR IGG4-RELATED DISEASE
(1) Marked lymphocyte and plasmacyte infiltration and fibrosis
(2) Infiltration of IgG4+ plasma cells: ratio of IgG4+/IgG+ cells >40% and >10 IgG4+ plasma cells/HPF
Definite: 1 + 2 + 3
Probable: 1 + 3
Possible: 1 + 2
owever, it is important to differentiate IgG4-RD from malignant tumours of each organ (e.g., cancer, lymphoma) and similar diseases (e.g., Sjögren syndrome, primary sclerosing cholangitis, Castleman disease, secondary retroperitoneal fibrosis, Wegener granulomatosis, sarcoidosis, Churg-Strauss syndrome) by additional histopathologic examination. Even when patients cannot be diagnosed using the CCD criteria, they may be diagnosed using organ-specific diagnostic criteria for IgG4-RD.
Reference: Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22(1):21-30.
22. OSTEOARTHRITIS
1986 American College of Rheumatology Criteria for the Classification of Knee Osteoarthritis
Clinical and laboratory Clinical and radiographic Clinical* Knee pain + at least five of the following: Knee pain + at least five of the following: Knee pain + at least three of six of the following: Age >50 years
Stiffness <30 minutes
Crepitus
Bony tenderness
Bony enlargement
No palpable warmth
ESR ≤40 mm/hr
RF <1:40
SF signs of OAAge >50 years
Stiffness <30 minutes
Crepitus
+ OsteophytesAge >50 years
Stiffness <30 minutes
Crepitus
Bony tenderness
Bony enlargement
No palpable warmthAlternate for the clinical category would be 4 out of 6.
Reference: Altman R, Asch E, Bloch D, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the knee. Arthritis Rheum. 1986;29:1039-1049.
1991 AMERICAN COLLEGE OF RHEUMATOLOGY CRITERIA FOR THE CLASSIFICATION OF OSTEOARTHRITIS OF THE HIP
Hip pain and at least two of the following three features:
Reference: Altman R, Alarcon G, Appelrouth D, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum. 1991;34:505-514.
1990 AMERICAN COLLEGE OF RHEUMATOLOGY CLASSIFICATION CRITERIA FOR OSTEOARTHRITIS OF THE HAND
Hand pain, aching, or stiffness and three or four of the following features:
* The 10 selected joints are the second and third distal interphalangeal (DIP), the second and third proximal interphalangeal, and the first carpometacarpal joints of both hands.
Reference: Altman R, Alarcón G, Appelrouth D, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hand. Arthritis Rheum. 1990;33:1601-1610.
23. GOUT
American College of Rheumatology/European League Against Rheumatism Gout Classification Criteria
Categories Score Step 1 Entry criterion (only apply criteria below to those meeting this entry criterion) At least 1 episode of swelling, pain, or tenderness in a peripheral joint or bursa Step 2 Sufficient criterion (if met, can classify as gout without applying criteria below) Presence of MSU crystals in a symptomatic joint or bursa (i.e., in synovial fluid) or tophus Step 3 Criteria (to be used if sufficient criterion not met) Pattern of joint or bursa involvement during symptomatic episode(s) ever† Ankle or midfoot (as part of monoarticular or oligoarticular episode without involvement of the first metatarsophalangeal joint)
Involvement of the first metatarsophalangeal joint (as part of monoarticular or oligoarticular episode)1
2Characteristics of symptomatic episode(s) ever:
Erythema overlying affected joint (patient reported or physician observed)
Can’t bear touch or pressure to affected joint
Great difficulty with walking or inability to use affected jointOne characteristic
Two characteristics
Three characteristics1
2
3Time course of episode(s) ever:
Presence (ever) of ≥2, irrespective of anti-inflammatory treatment
Time to maximal pain <24 hours
Resolution of symptoms in ≤14 days
Complete resolution (to baseline level) between symptomatic episodesOne typical episode
Recurrent typical episodes1
2Clinical evidence of tophus:
Draining or chalklike subcutaneous nodule under transparent skin, often with overlying vascularity, located in typical locations: joints, ears, olecranon bursae, finger pads, tendons (e.g., Achilles tendon)Present 4 Laboratory Serum urate: Measured by uricase method. Ideally should be scored at a time when the patient was not receiving urate-lowering treatment and it was >4 weeks from the start of an episode (i.e., during intercritical period); if practicable, retest under those conditions. The highest value irrespective of timing should be scored. <4 mg/dL (<0.24 mmol/L)‡
6–8 mg/dL (0.36–<0.48 mmol/L)
8– <10 mg/dL (0.48–<0.60 mmol/L)
≥10 mg/dL (≥0.60 mmol/L)-4
2
3
4Synovial fluid analysis of a symptomatic (ever) joint or bursa (should be assessed by a trained observer) § MSU negative -2 Imaging Imaging evidence of urate deposition in symptomatic (ever) joint or bursa: ultrasound evidence of double-contour sign¶ or DECT demonstrating urate deposition* Present (either modality) 4 Imaging evidence of gout-related joint damage: conventional radiography of the hands and/or feet demonstrates at least 1 erosion** Present 4 §-If polarizing microscopy of synovial fluid from a symptomatic (ever) joint or bursa by a trained examiner fails to show monosodium urate monohydrate (MSU) crystals, subtract 2 points. If synovial fluid was not assessed, score this item as 0.
¶- Hyperechoic irregular enhancement over the surface of the hyaline cartilage that is independent of the insonation angle of the ultrasound beam (note: false-positive double-contour sign [artefact] may appear at the cartilage surface but should disappear with a change in the insonation angle of the probe).
*- The presence of color-coded urate at articular or periarticular sites. Images should be acquired using a dual-energy computed tomography (DECT) scanner, with data acquired at 80 and 140 kV and analyzed using gout-specific software with a two-material decomposition algorithm that colour codes urate. A positive scan is defined as the presence of color-coded urate at articular or periarticular sites. Nailbed, submillimetre, skin, motion, beam hardening, and vascular artefacts should not be interpreted as DECT evidence of urate deposition.
**- Erosion is defined as a cortical break with sclerotic margin and overhanging edge, excluding distal interphalangeal joints and gull’s wing appearance.
Reference: Neogi T, Jansen TL, Dalbeth N, et al. 2015 Gout Classification Criteria: an American College of Rheumatology /European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2015;67(10):2557-2568.
24. FIBROMYALGIA
1990 AMERICAN COLLEGE OF RHEUMATOLOGY CRITERIA FOR THE CLASSIFICATION OF FIBROMYALGIA
1. History of widespread pain
Definition: Pain is considered widespread when all of the following are present: pain in the left side of the body, pain in the right side of the body, pain above the waist, and pain below the waist. In addition, axial skeletal pain (cervical spine or anterior chest or thoracic spine or low back) must be present. In this definition, shoulder and buttock pain is considered as pain for each involved side. “Low back” pain is considered lower segment pain.
2. Pain in 11 of 18 tender point sites on digital palpation
Definition: Pain, on digital palpation, must be present in at least 11 of the following 18 sites:
Occiput: bilateral, at the suboccipital muscle insertions
Low cervical: bilateral, at the anterior aspects of the intertransverse spaces at C5–C7
Trapezius: bilateral, at the midpoint of the upper border
Supraspinatus: bilateral, at origins, above the scapula spine near the medial border
Second rib: bilateral, at the second costochondral junctions, just lateral to the junctions on upper surfaces
Lateral epicondyle: bilateral, 2 cm distal to the epicondyles
Gluteal: bilateral, in upper outer quadrants of buttocks in anterior fold of muscle
Greater trochanter: bilateral, posterior to the trochanteric prominence
Knee: bilateral, at the medial fat pad proximal to the joint line
Digital palpation should be performed with an approximate force of 4 kg.
For a tender point to be considered “positive,” the subject must state that the palpation was painful. “Tender” is not to be considered “painful.”
For classification purposes, patients will be said to have fibromyalgia if both criteria are satisfied. Widespread pain must have been present for at least 3 months.
The presence of a second clinical disorder does not exclude the diagnosis of fibromyalgia.
Reference: Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia: report of the multicentre criteria committee. Arthritis Rheum. 1990;33:160-172.
2010 AMERICAN COLLEGE OF RHEUMATOLOGY PRELIMINARY CRITERIA FOR THE DIAGNOSIS OF FIBROMYALGIA
Criteria
A patient satisfies diagnostic criteria for fibromyalgia if the following three conditions are met:
Ascertainment
1. WPI: Note the number of areas in which the patient has had pain over the past week. In how many areas has the patient had pain? Score will be between 0 and 19.
10. SS scale score:
*- Somatic symptoms that might be considered: muscle pain, irritable bowel syndrome, fatigue or tiredness, thinking or remembering problem, muscle weakness, headache, pain or cramps in the abdomen, numbness or tingling, dizziness, insomnia, depression, constipation, pain in the upper abdomen, nausea, nervousness, chest pain, blurred vision, fever, diarrhoea, dry mouth, itching, wheezing, Raynaud phenomenon, hives or welts, ringing in ears, vomiting, heartburn, oral ulcers, loss of or change in taste, seizures, dry eyes, shortness of breath, loss of appetite, rash, sun sensitivity, hearing difficulties, easy bruising, hair loss, frequent urination, painful urination, and bladder spasms.
Reference: Wolfe F, Clauw DJ, Fitzcharles MA, et al. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res (Hoboken). 2010;62(5):600-610.
2016 Revisions to the 2010/2011 Fibromyalgia Diagnostic Criteria
“A diagnosis of fibromyalgia is valid irrespective of other diagnoses. A diagnosis of fibromyalgia does not exclude the presence of other clinically important illnesses.”
Criteria A patient satisfies modified 2016 fibromyalgia criteria if the following 3 conditions are met:
Ascertainment (1) WPI: note the number of areas in which the patient has had pain over the last week. In how many areas has the patient had pain? Score will be between 0 and 19
Region Areas Score Left Upper Jaw, shoulder girdle, Upper arm, lower arm 0-4 Right Upper Jaw, shoulder girdle, Upper arm, lower arm 0-4 Axial Neck, Upper back, lower back, upper chest, lower chest 0-5 Left Lower Hip, Upper thigh and Lower thigh 0-3 Right Lower Hip, Upper thigh and Lower thigh 0-3 (2) Symptom severity scale (SSS) score
For the each of the 3 symptoms above, indicate the level of severity over the past week using the following scale:
0 = No problem
1 = Slight or mild problems, generally mild or intermittent
2 = Moderate, considerable problems, often present and/or at a moderate level
3 = Severe: pervasive, continuous, life-disturbing problems
The symptom severity scale (SSS) score: is the sum of the severity scores of the 3 symptoms (fatigue, waking unrefreshed, and cognitive symptoms) (0–9) plus the sum (0–3) of the number of the following symptoms the patient has been bothered by that occurred during the previous 6 months:
(1) Headaches (0–1) (2) Pain or cramps in lower abdomen (0–1) (3) And depression (0–1)
The final symptom severity score is between 0 and 12 The fibromyalgia severity (FS) scale is the sum of the WPI and SSS. The FS scale is also known as the polysymptomatic distress (PSD) scale.
Wolfe F, Clauw DJ, FitzCharles M, Goldenerberg D, Häuser W, Katz RS, Russell IJ, Mease PJ, Russell A, Walitt B. 2016 Revisions to the 2010/2011 Fibromyalgia Diagnostic Criteria [abstract]. Arthritis Rheumatol. 2016; 68 (suppl 10). https://acrabstracts.org/abstract/2016-revisions-to-the-20102011-fibromyalgia-diagnostic-criteria/. Accessed March 21, 2019.
25. HYPERMOBILITY (Benign Joint Hypermobility Syndrome)
BRIGHTON CRITERIA
Major criteria
Minor criteria
Requirement for diagnosis
Any one of the following:
1- Beighton score
Score Component Left Right 1. Passive dorsiflexion and hyperextension of the fifth MCP joint beyond 90° 1 1 2. Passive apposition of the thumb to the flexor aspect of the forearm 1 1 3. Passive hyperextension of the elbow beyond 10° 1 1 4. Passive hyperextension of the knee beyond 10° 1 1 5. Active forward flexion of the trunk with the knees fully extended so that the palms of the hands rest flat on the floor 1 Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 196951(3):444-4453.
Reference: Grahame R. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27:1777-1779.
26. COMPLEX REGIONAL PAIN SYNDROME
INTERNATIONAL ASSOCIATION FOR THE STUDY OF PAIN 2007 PROPOSED DIAGNOSTIC CRITERIA FOR COMPLEX REGIONAL PAIN SYNDROME - Budapest consensus criteria
General definition of the syndrome
CRPS describes an array of painful conditions that are characterized by a continuing (spontaneous, evoked, or both) regional pain that is seemingly disproportionate in time or degree to the usual course of any known trauma or other lesion. The pain is regional (not in a specific nerve territory or dermatome) and usually has a distal predominance of abnormal sensory, motor, sudomotor, vasomotor, or trophic findings. The syndrome shows variable progression over time.
To make the clinical diagnosis, the following criteria must be met:
1. Continuing pain that is disproportionate to any inciting event
2. Must report at least one symptom in three of the four following categories:
3. Must display at least one sign at time of evaluation in two or more of the following categories:
4. There is no other diagnosis that better explains the signs and symptoms
For research purposes, the diagnostic decision rule should be at least one symptom in all four symptom categories and at least one sign (observed at evaluation) in two or more sign categories.
Reference: Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med. 2007;8(4):326-331.
27. DIFFUSE IDIOPATHIC SKELETAL HYPEROSTOSIS
Criteria of Resnick and Niwayama
Reference: Resnick D, Niwayama G. Radiographic and pathologic features of spinal involvement in diffuse idiopathic skeletal hyperostosis (DISH). Radiology, 1976;119:559.
Criteria of Utsinger
Reference: Utsinger PD. Diffuse idiopathic skeletal hyperostosis. Clin Rheum Dis. 1985;11:325.
-
- Symmetric proximal muscle weakness
- Muscle biopsy evidence of myositis
- Elevated serum muscle enzymes
- Myopathic changes on electromyography
- Typical rash of DM (a distinguishing feature for DM and PM)
- Duration >6 months
- Age of onset >30 years
- Weakness
- Finger flexor weakness
- Wrist flexor >extensor weakness
- Quadriceps weakness ≤grade 4 MRC
- Inflammatory myopathy (with partial invasion)
- Rimmed vacuoles
- Either
- Intracellular amyloid deposits,
- 15- to 18-nm tubulofilaments by EM
- 1. Age at disease onset ≥50 years: development of symptoms or findings beginning at age 50 or older
- 2. New headache: new onset of or new type of localized pain in the head
- 3. Temporal artery abnormality: temporal artery tenderness to palpation or decreased pulsation, unrelated to arteriosclerosis of cervical arteries
- 4. ESR rate: ESR ≥50 mm/hr by the Westergren method
- 5. Abnormal artery biopsy: biopsy specimen with artery showing vasculitis characterized by a predominance of mononuclear cell infiltration or granulomatous inflammation, usually with multinucleated giant cells
- 1. Erythema and cracking of lips, strawberry tongue, or erythema of the oral and pharyngeal mucosa
- 2. Bilateral bulbar conjunctival injection without exudate
- 3. Rash: maculopapular, diffuse erythroderma, or erythema multiforme–like
- 4. Erythema and edema of the hands and feet in the acute phase or periungual desquamation in the subacute phase
- 5. Cervical lymphadenopathy (≥1.5-cm diameter), usually unilatera
- Changes in peripheral extremities or perineal area
- Polymorphous exanthema
- Bilateral conjunctival injection
- Changes of lips and oral cavity: injection of oral and pharyngeal mucosa
- Cervical lymphadenopathy
- Buccal ulceration
- Genital ulceration
- Eye lesions
- Skin lesions
- Gastrointestinal lesions
- Thrombophlebitis
- Cardiovascular lesions
- Arthritis
- Central nervous system lesions
- Family history of Behçet disease
- Do you remember one or more episodes of small red spots on your skin, particularly involving the lower limbs?
- Have you ever had red spots on your lower extremities, which leave a brownish colour after their disappearance?
- Has a doctor ever told you that you have viral hepatitis?
- Reduced serum C4
- Positive serum rheumatoid factor
- Positive serum monoclonal component
- 1. Nasal or oral inflammation: development of painful or painless oral ulcers or purulent or bloody nasal discharge
- 2. Abnormal chest radiograph: chest radiograph showing the presence of nodules, fixed infiltrates, or cavities
- 3. Urinary sediment: microhematuria (>5 RBCs per high-power field) or RBC casts in urine sediment
- 4. Granulomatous inflammation on biopsy: histologic changes showing granulomatous inflammation within the wall of an artery or in the perivascular or extravascular area (artery or arteriole)
- Weight loss of 4 kg or more
- Livedo reticularis
- Testicular pain/tenderness
- Myalgia or leg weakness/tenderness
- Mononeuropathy or polyneuropathy
- Diastolic blood pressure greater than 90 mm/Hg
- Elevated blood urea nitrogen (BUN) or creatinine level unrelated to dehydration or obstruction
- Presence of hepatitis B surface antigen or antibody in serum
- Arteriogram demonstrating aneurysms or occlusions of the visceral arteries
- Presence of polymorphonuclear neutrophils in a biopsy specimen from a small- or medium-sized artery
- Fever >39°C, intermittent, ≥1 week
- Arthralgia ≥2 weeks
- Characteristic rash
- WBC count >10,000/mL (>80% granulocytes)
- Sore throat
- Lymphadenopathy or splenomegaly
- LFT abnormal
- Negative ANA/RF
- Infections
- Malignancies
- Rheumatic diseases
- Spiking fever ≥39°C
- Arthralgia
- Transient erythema
- Pharyngitis
- PMN leukocytes ≥80%
- Glycosylated ferritin ≤20%
- Maculopapular rash
- WBC count >10,000/mL
- 1. Recurrent chondritis of both auricles
- 2. Nonerosive inflammatory polyarthritis
- 3. Chondritis of nasal cartilages
- 4. Inflammation of ocular structures
- 5. Chondritis of respiratory tract
- 6. Cochlear or vestibular damage
- 1. Three of six McAdam et al’s criteria
- 2. One of six McAdam et al’s criteria and a positive histologic confirmation
- 3. Two of six McAdam et al’s criteria and response to corticosteroid or dapsone
- Proven inflammation in two of three cartilages: auricular, nasal, and laryngotracheal
- Proven inflammation in one of the above and meeting two other signs from ocular inflammation, hearing loss, vestibular dysfunction, or seronegative inflammatory arthritis
- 1) McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polytonicities: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- 2) Damiani JM, Levine HL. Relapsing polytonicities. Report of ten cases. Laryngoscope. 1979;89: 929-46.
- 3) Michet CJ, McKenna CH, Luthra HS, O’Fallon WM. Relapsing polytonicities: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- 1. Clinical examination showing characteristic diffuse or localized swelling or masses in single or multiple organs
- 2. Hematologic examination shows elevated serum IgG4 concentrations (≥135 mg/dL)
- 3. Histopathologic examination shows:
- ESR <20 mm/hr
- Radiographic femoral or acetabular osteophytes
- Radiographic joint space narrowing (superior, axial, or medial)
- Hard tissue enlargement of two or more of 10 selected joints
- Hard tissue enlargement of two or more DIP joints
- Fewer than three swollen MCP joints
- Deformity of at least one of 10 selected joints*
- 1. Widespread pain index (WPI) 7 and symptom severity (SS) scale score 5 or WPI 3–6 and SS scale score 9
- 2. Symptoms have been present at a similar level for at least 3 months.
- 3. The patient does not have a disorder that would otherwise explain the pain.
- Shoulder girdle, left, Shoulder girdle, right
- Upper arm, left, Upper arm, right
- Lower arm, left, Lower arm, right
- Hip (buttock, trochanter), left, Hip (buttock, trochanter), right
- Upper leg, left, Upper leg, right
- Lower leg, left, Lower leg, right
- Jaw, left, Jaw, right
- Chest, Abdomen
- Upper back, Lower back, Neck
- Fatigue
- Waking unrefreshed
- Cognitive symptoms
- For the each of the 3 symptoms above, indicate the level of severity over the past week using the following scale:
- 0 = no problem
- 1 = slight or mild problems, generally mild or intermittent
- 2 = moderate, considerable problems, often present or at a moderate level
- 3 = severe: pervasive, continuous, life-disturbing problems
- Considering somatic symptoms in general, indicate whether the patient has:*
- 0 = no symptoms
- 1 = few symptoms
- 2 = a moderate number of symptoms
- 3 = a great deal of symptoms
- The SS scale score is the sum of the severity of the three symptoms (fatigue, waking unrefreshed, cognitive symptoms)
- plus the extent (severity) of somatic symptoms in general. The final score is between 0 and 12.
- (1) Widespread pain index (WPI) ³ 7 and symptom severity scale (SSS) score ³5 OR WPI of 4–6 and SSS score ³ 9.
- (2) Generalized pain, defined as pain in at least 4 of 5 regions, must be present. Jaw, chest, and abdominal pain are not included in generalized pain definition.
- (3) Symptoms have been generally present for at least 3 months.
- 4) A diagnosis of fibromyalgia is valid irrespective of other diagnoses. A diagnosis of fibromyalgia does not exclude the presence of other clinically important illnesses.
- a) Fatigue
- b) Waking unrefreshed
- c) Cognitive symptoms
- Beighton score of ≥41
- Arthralgia for longer than 3 months in four or more months
- Beighton score1 of 1, 2, or 3
- Arthralgia (>3-month duration) in one to three joints or back pain (>3-month duration) or spondylosis, spondylolysis, or spondylolisthesis
- Dislocation or subluxation in more than one joint or in one joint on more than one occasion
- Three or more soft tissue lesions (e.g., epicondylitis, tenosynovitis, bursitis)
- Marfanoid habitus: tall, slim, span greater than height (>1.03 ratio), upper segment less than lower segment (<0.89 ratio), arachnodactyly
- Skin striae, hyperextensibility, thin skin, or abnormal scarring
- Ocular signs: drooping eyelids, myopia, antimongoloid slant
- Varicose veins, hernia, or uterine or rectal prolapse
- Mitral valve prolapse
- Two major criteria
- One major plus two minor criteria
- Four minor criteria
- Two minor criteria and unequivocally affected first-degree relative in family history
- Sensory: Reports of hyperesthesia or allodynia
- Vasomotor: Reports of temperature asymmetry, skin colour changes, or skin colour asymmetry
- Sudomotor or edema: Reports of edema, sweating changes, or sweating asymmetry
- Motor or trophic: Reports of decreased range of motion, motor dysfunction (weakness, tremor, dystonia), or trophic changes (hair, nail, skin)
- Sensory: Evidence of hyperalgesia (to pinprick), allodynia (to light touch, temperature sensation, deep somatic pressure, or joint movement)
- Vasomotor: Evidence of temperature asymmetry (>1°C), skin colour changes, or asymmetry
- Sudomotor or edema: Evidence of edema, sweating changes, or sweating asymmetry
- Motor or trophic: Evidence of decreased range of motion, motor dysfunction (weakness, tremor, dystonia), or trophic changes (hair, nail, skin)
- 1. The presence of flowing calcification and ossification along the anterolateral aspect of at least four contiguous vertebral bodies with or without associated localized pointed excrescences at the intervening vertebral body–intervertebral disk junctions
- 2. The presence of relative preservation of intervertebral disk height in the involved vertebral segment and the absence of extensive radiographic changes of “degenerative” disk disease, including vacuum phenomena and vertebral body marginal sclerosis
- 3. The absence of apophyseal joint bony ankylosis and sacroiliac joint erosion, sclerosis, or intraarticular osseous fusion
- 1. Continuous ossification along the anterolateral aspect of at least four contiguous vertebral bodies, primarily in the thoracolumbar spine. Ossification begins as a fine, ribbon-like wave of bone but commonly develops into a broad, bumpy, buttress-like band of bone.
- 2. Continuous ossification along the anterolateral aspect of at least two contiguous vertebral bodies
- 3. Symmetric and peripheral enthesopathy involving the posterior heel, superior patella, or olecranon, with the entheseal new bone having a well-defined cortical margin.
-
1. JUVENILE IDIOPATHIC ARTHRITIS
International League of Associations for Rheumatology Classification for Juvenile Idiopathic Arthritis
| JIA Category | Inclusion criteria | Exclusion criteria |
|---|---|---|
| Systemic arthritis | Arthritis* in one or more joints with or preceded by fever of at least 2 weeks’ duration documented to be daily for at least 3 days and accompanied by one or more of evanescent erythematous rash, lymphadenopathy, hepatomegaly or splenomegaly (or both), or serositis | Psoriasis or a history of psoriasis in the patient or a first-degree relative Arthritis in an HLA-B27–positive male beginning after the sixth birthday AS, ERA, sacroiliitis with IBD, reactive arthritis or acute anterior uveitis, or a history of one of these disorders in a first-degree relative Presence of IgM RF on at least two occasions at least 3 months apart |
| Oligoarthritis (persistent or extended) | Arthritis in one to four joints in the first 6 months Persistent disease affects no more than four joints throughout the course of the disease, but extended disease affects a total of more than four joints after the first 6 months | Psoriasis or a history of psoriasis in the patient or a first-degree relative Arthritis in an HLA-B27–positive male beginning after the sixth birthday AS, ERA, sacroiliitis with IBD, reactive arthritis or acute anterior uveitis, or a history of one of these disorders in a first-degree relative Presence of IgM RF on at least two occasions at least 3 months apart Systemic JIA in the patient |
| Polyarthritis RF- | Arthritis affecting five or more joints in the first 6 months of disease. A test for RF is negative. | Psoriasis or a history of psoriasis in the patient or a first-degree relative Arthritis in an HLA-B27–positive male beginning after the sixth birthday AS, ERA, sacroiliitis with IBD, reactive arthritis or acute anterior uveitis, or a history of one of these disorders in a first-degree relative Presence of IgM RF on at least two occasions at least 3 months apart Systemic JIA in the patient |
| Polyarthritis RF+ | Arthritis affecting five or more joints in the first 6 months of disease | Psoriasis or a history of psoriasis in the patient or a first-degree relative Arthritis in an HLA-B27–positive male beginning after the sixth birthday AS, ERA, sacroiliitis with IBD, reactive arthritis or acute anterior uveitis, or a history of one of these disorders in a first-degree relative Systemic JIA in the patient |
(cont.)
| Psoriatic arthritis | Arthritis plus psoriasis or Arthritis plus at least two of the following: dactylitis, nail pitting or onycholysis, psoriasis in a first-degree relative | Arthritis in an HLA-B27–positive male beginning after the sixth birthday AS, ERA, sacroiliitis with IBD, reactive arthritis or acute anterior uveitis, or a history of one of these disorders in a first-degree relative Presence of IgM RF on at least two occasions at least 3 months apart Systemic JIA in the patient |
|---|---|---|
| Enthesitis-related arthritis | Arthritis plus enthesitis or Arthritis or enthesitis plus at least two of the following: presence of or a history of sacroiliac joint tenderness or inflammatory lumbosacral pain, presence of HLA-B27 antigen, onset of arthritis in a male older than 6 years of age, acute (symptomatic) anterior uveitis, history of AS, ERA, sacroiliitis with IBD, reactive arthritis, or acute anterior uveitis in a first-degree relative | Psoriasis or a history of psoriasis in the patient or a first-degree relative Presence of IgM RF on at least two occasions at least 3 months apart Systemic JIA in the patient |
| Undifferentiated arthritis | Arthritis that fulfils criteria in none of the above categories or fulfils criteria in two or more of the above categories | NA |
*JIA is arthritis of unknown etiology that begins before the patient’s 16th birthday and persists for at least 6 weeks.
Inflammatory lumbosacral pain is defined as lumbosacral pain at rest with morning stiffness that improves with movement.
Reference: Colbert RA. Classification of juvenile spondyloarthritis: enthesitis-related arthritis and beyond. Nat Rev Rheumatol. 2010;6(8):477-485.
2. Childhood Takayasu arteritis
Final European League Against Rheumatism/Paediatric Rheumatology International Trials Organisation/Paediatric Rheumatology European Society Childhood Takayasu Arteritis Criteria (with Glossary) and Classification Definition
| Criterion | Definition |
|---|---|
| Angiographic abnormality (mandatory criterion) | Angiography (conventional, CT, or MRI) of the aorta or its main branches and pulmonary arteries showing aneurysm or dilation, narrowing, occlusion, or thickened arterial wall not caused by fibromuscular dysplasia or similar causes; changes usually focal or segmental |
| 1. Pulse deficit or claudication | Lost, decreased, or unequal peripheral artery pulse(s) Claudication: focal muscle pain induced by physical activity |
| 2. BP discrepancy | Discrepancy of four-limb systolic BP >10 mm Hg difference in any limb |
| 3. Bruits | Audible murmurs or palpable thrills over large arteries |
| 4. Hypertension | Systolic or diastolic BP >95th percentile for height |
| 5. Acute phase reactant | ESR >20 mm per first hour or CRP any value above normal (according to the local laboratory) |
c-TA EULAR/PRINTO/ PRES Ankara 2008 classification definition-
Angiographic abnormalities of the aorta or its main branches and pulmonary arteries showing aneurysm or dilation (mandatory criterion) plus one of the five following criteria:
Pulse deficit or claudication, Four-limb BP discrepancy, Bruits, Hypertension, Acute phase reactant
Reference: Ozen S, Pistorio A, Iusan SM, et al; Paediatric Rheumatology International Trials Organisation (PRINTO). EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798-806.
3. Childhood Wegener Granulomatosis
Final European League Against Rheumatism/Paediatric Rheumatology International Trials Organisation/Paediatric Rheumatology European Society Childhood Wegener Granulomatosis Criteria (with Glossary) and Classification Definition
| Criterion | Glossary |
|---|---|
| Histopathology | Granulomatous inflammation within the wall of an artery or in the perivascular or extravascular area |
| Upper airway involvement | Chronic purulent or bloody nasal discharge or recurrent epistaxis, crusts, or granulomata Nasal septum perforation or saddle nose deformity Chronic or recurrent sinus inflammation |
| Laryngo-tracheo-bronchial involvement | Subglottic, tracheal, or bronchial stenosis |
| Pulmonary involvement | Chest radiography or CT showing the presence of nodules, cavities or fixed infiltrates |
| ANCA | ANCA positivity by immunofluorescence or by ELISA (MPO/p or PR3/c ANCA) |
| Renal involvement | Proteinuria >0.3 g/24 hr or >30 mmol/mg of urine albumin/creatinine ratio on a spot morning sample Haematuria or RBC casts: >5 RBCs/high-power field or RBC casts in the urinary sediment or ≥2+ on dipstick Necrotizing pauci-immune glomerulonephritis |
c-WG EULAR/PRINTO/PRES Ankara 2008 classification definition: At least three of the six following criteria
| Area involvement | Descriptors |
|---|---|
| Upper airway involvement | Chronic purulent or bloody nasal discharge, or recurrent epistaxis/crusts/granulomata Nasal septal perforation or saddle-nose deformity Chronic or recurrent sinus inflammation |
| Pulmonary involvement | Chest X-ray or CT scan showing the presence of nodules, cavities, or fixed infiltrates |
| Renal involvement | Proteinuria >0.3 g/24 hours or greater than 30 mmol/mg of urine albumin/creatinine ratio on a spot morning sample Hematuria or red blood cell casts: >5 red blood cells per high-power field, or red blood cell casts in urinary sediment, or >2+ on dipstick Necrotizing pauci-immune glomerulonephritis |
| Granulomatous inflammation | Granulomatous inflammation within wall of artery or in perivascular or extravascular area of artery or arteriole |
| Laryngo tracheo bronchial stenosis | Subglottic, tracheal, or bronchial stenosis |
| ANCA positivity | ANCA positivity by immunofluorescence or by ELISA (MPO/p or PR3/c ANCA) |
Reference: Ozen S, Pistorio A, Iusan SM, et al; Paediatric Rheumatology International Trials Organisation (PRINTO). EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798-806.
4. Uveitis
Standardization of Uveitis Nomenclature.
| Type | Primary Site of Inflammation | Includes |
|---|---|---|
| Anterior Chamber | Iritis | |
| Intermediate | Vitreous | Pars planitis Posterior cyclitis Hyalitis |
| Posterior | Retina or Choroid | Focal, multifocal, or diffuse choroiditis Chorioretinitis Retinochoroiditis Retinitis Neuroretinitis |
5. Juvenile Systemic Sclerosis
Preliminary Classification Criteria for Juvenile Systemic Sclerosis
| Criteria | Organ | Features |
|---|---|---|
| Major | Sclerosis/induration of skin proximal to MCP | |
| Minor | Skin | Sclerodactyly |
| Vascular | Raynaud phenomenon Nailfold capillary abnormalities Digital tip ulcers | |
| Gastrointestinal | Dysphagia Gastroesophageal reflux |
|
| Renal | Renal crisis New-onset arterial hypertension |
|
| Cardiac | Arrhythmias Heart failure |
|
| Musculoskeletal | Pulmonary fibrosis (HRCT/X-ray) DLCO Pulmonary hypertension |
|
| Respiratory | Tendon friction rubs Arthritis Myositis |
|
| Neurological | Neuropathy Carpal tunnel syndrome |
|
| Serology | Antinuclear antibodies SSc selective autoantibodies (anticentromere, anti-topoisomerase I, anti-fibrillarin, anti-PM-Scl, anti-fibrillin or anti-RNA polymerase I or III) |
PRES/ACR/EULAR Ad Hoc Committee on Classification Criteria for JSSc (2007). The Pediatric Rheumatology European Society/American College of Rheumatology/European League Against Rheumatism. Provisional Classification Criteria for Juvenile Systemic Sclerosis. Arthritis Rheum 57(2): 203–212. *A patient, aged less than 16 years, shall be classified as having juvenile systemic sclerosis if the one major and at least two of the 20 minor criteria are present. This set of classification criteria have a sensitivity of 90%, and a specificity of 96%.
6. Juvenile Primary Sjogren Syndrome
Proposed Criteria for Juvenile Primary Sjogren Syndrome
I. Clinical symptoms
- 1. Oral (dry mouth, recurrent parotitis, or enlargement of parotid glands)
- 2. Ocular (recurrent conjunctivitis without obvious allergic or infectious etiology, keratoconjunctivitis sicca)
- 3. Other mucosal involvement (recurrent vaginitis)
- 4. Systemic (fever of unknown origin, noninflammatory arthralgias, hypokalemic paralysis, abdominal pain)
II. Immunological abnormalities (presence of at least one of the following:
- anti-SSA, anti-SSB, high-titer ANA, RF)
III. Other laboratory abnormalities or additional investigations
- 1. Biochemical (elevated serum amylase)
- 2. Hematological (leucopenia, high ESR)
- 3. Immunological (polyclonal hyperimmunoglobulinemia)
- 4. Nephrological (renal tubular acidosis)
- 5. Histological proof of lymphocytic infiltration of salivary glands or other organs
- 6. Objective documentation of ocular dryness (Bengal red staining, Schirmer test)
- 7. Objective documentation of parotid gland involvement (sialography)
IV. Exclusion of all other autoimmune diseases
- Presence of four or more criteria required for diagnosis
- From Bartunkova, Sediva, Vencovsky, et al., Primary Sjögren’s syndrome in children and adolescents: proposal for diagnostic criteria,
- Clin. Exp. Rheumatol. 17 (1999) 381–386.
7. EULAR/PReS Classification of Childhood Vasculitis
| i. Predominantly large-vessel vasculitis | Takayasu Arteritis |
|---|---|
| ii. Predominantly medium-sized vessel vasculitis | Childhood polyarteritis nodosa Cutaneous polyarteritis Kawasaki disease |
| iii. Predominantly small-sized vessel vasculitis | A. Granulomatous Wegener granulomatosis Churg–Strauss syndrome B. Nongranulomatous Microscopic polyangiitis Henoch–Schönlein purpura Isolated cutaneous leukocytoclastic vasculitis Hypocomplementemic urticarial vasculitis |
| iv. Other vasculitides | Bechet disease Vasculitis secondary to infection (including hepatitis B-associated polyarteritis nodosa), malignancies, and drugs (including hypersensitivity vasculitis) Vasculitis associated with connective tissue diseases Isolated vasculitis of the central nervous system Cogan syndrome Unclassified |
S.Ozen,N. Ruperto, M. Dillon, et al., EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides, Ann. Rheum. Dis. 65 (2006) 936–941.
8. Childhood Polyarteritis Nodosa
Classification Criteriafor Childhood Polyarteritis Nodosa
Evidence of necrotizing vasculitis in medium or small arteries or an angiographic abnormality showing aneurysm, stenosis, or occlusion of a medium- or small-sized artery (histopathology or angiography mandatory),
plus, one out of five of the following criteria:
- 1.Skin involvement (livedo reticularis, skin nodules, or infarcts)
- 2. Myalgia or muscle tenderness
- 3. Hypertension (systolic/diastolic blood pressure greater than 95th percentile for height)
- 4. Peripheral neuropathy (sensory peripheral neuropathy or motor mononeuritis multiplex)
- 5. Renal involvement (proteinuria >0.3 g/24 hours or >30 mmol/mg of urine albumin/creatinine ratio on a spot morning sample; hematuria or red blood cell casts, >5 red blood cells/high-power field, red blood cell casts in the urinary sediment, or equal to 2+ on dipstick; or impaired renal function, measured or calculated glomerular filtration rate [Schwartz formula] <50% normal)
Adapted from S. Ozen, A. Pistorio, S.M. Iusan, et al., EULAR/PRINTO/PRES criteria for Henoch Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria, Ann. Rheum. Dis. 69 (2010) 798–806.
9.Childhood Primary Angiitis of CNS
Proposed DiagnosticCriteria for cPACNS
| A newly acquired neurological and/or psychiatric deficit |
|---|
| PLUS |
| Angiographic and/or histological evidence of CNS vasculitis in the absence of any systemic condition known to be associated with or mimic CNS vasculitis. |
Prepared by
Dr Avinash Jain
SGPGI, Lucknow
Dr Rutviz Mistry
SGPGI, Lucknow
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma, Dr Praveen Hisaria and Dr Sapan C Pandya
Infections are the major cause of morbidity and mortality in patients with Autoimmune Rheumatic Diseases. Immune response in these patients is impaired and the “immunosuppressive” medications used to treat them add fuel to the fire. Infections are the biggest threat in the management of rheumatic conditions. Influenza, invasive pneumococcal infection, herpes zoster and hepatitis B are the major vaccine preventable infections seen in our patients.
Vaccination in rheumatic disease and its studies present with a unique set of challenges. Vaccination leads to immune response to particular antigen; however, non-specific response in this situation might lead to flare of the autoimmune disease. Ideally, studies on efficacy should use clinical endpoints to test the role of vaccines in rheumatic diseases to establish its clinical benefits. Such studies with clinical endpoints are logistically demanding and require a large sample size. Hence, most of the current studies use laboratory parameters (serologic titres of antibodies or T cell reactivity against antigen) to establish the efficacy of the vaccines. However, laboratory surrogates lack correlation with the clinical endpoints of reducing infection. Studies related to vaccination need to be interpreted with this consideration in mind.
In this write-up we will focus on the evidence of efficacy of various vaccines in rheumatic diseases and end with the current recommendations pertaining to vaccination.
Influenza virus
Influenza vaccine currently available in the market include inactivated and live attenuated. Trivalent and tetravalent vaccines containing three and four strains are available; however, the most commonly used is the trivalent inactivated vaccine. Multiple studies including a prospective1, and a retrospective large registry based Taiwanese study2 with clinical endpoints suggest reduction in pneumonitis, bronchitis, hospitalization in Rheumatoid arthritis and SLE patients vaccinated with influenza vaccine as compared to unvaccinated patients. In SLE, the serological response to the influenza subunit varied among different studies. Few studies show mild reduction in sero-protection while the others did not show any difference in seroconversionbetween vaccinated and unvaccinated patients.3,4 Serologic evidence of protection in Systemic Sclerosis5, Granulomatosis with polyangitis6 and Sjögren’s syndrome7 have been observed after influenza vaccination. Significant body of evidence exists to suggest efficacy of Influenza vaccination with concomitant use of glucocorticoids, csDMARDs and antiTNF therapy and tocilizumab. In one study, the arm on combination therapy with methotrexate and anti-TNF had lower titres of antibodies to influenza as compared to methotrexate alone, however, multiple other studies have shown good response with combination therapy as well.8 Studies with rituximab in RA however have documented significant lower seroconversion rates.9None of the studies have raised concerns regarding the safety or flare of underlying autoimmune disease.
Antigenic Drift and Shift leads to changing immunogenicity of the Influenza strains each year. Depending on the strains in circulation in a particular demographic area, the manufacturers “update” their vaccine to include the recent strains. This should be kept in mind while administering vaccine to patients. The best time to vaccinate with yearly shot is before the onset of monsoon10 (April-May) since influenza infection is particularly more common in monsoon and winter.
Thus, current body of evidence suggest influenza vaccines are well tolerated but underutilized in rheumatic diseases patients and are generally immunogenic even with immunosuppressants with the exception of rituximab.Vaccines should ideally be administered before B cell-depleting biological therapy [BCDT] is started or, when patients are on such a treatment already, at least 6 months after the start but 4 weeks before the next course.The European League Against Rheumatism (EULAR) recommend yearly vaccination with influenza of all patients with rheumatic diseases11.
Streptococcus pneumoniae
Currently two forms of pneumococcal vaccines are available. PPSV23 is derived from polysaccharide capsule while PCV13 is a conjugated vaccine with diphtheria carrier protein. PPSV23 response is T cell independent while PCV13 is T cell dependent. The immunological response is robust in PCV13 compared to PPSV23.Hence, boosters are required in PPSV23 while a single dose is sufficient in PCV13. Majority of the available literature has used PPSV23 in rheumatic diseases. Another reason for heterogeneity among the available data is lack of generally accepted serologic protection criteria for immunologic response to Pneumococcal vaccine.
In RA, good body of evidence exists to suggest adequate serologic response to pneumococcal vaccination independent of the DMARD used and disease activity12. However, newer studies have documented mildly reduced seroconversion with methotrexate-antiTNF combination and severely impaired humoral response with BCDT.13,14 Recent studies of PPSV23 in SLE suggest reduced immunogenicity as compared to healthy controls.15 Efficacy of pneumococcal vaccine is also established in Systemic sclerosis16 and Psoriatic arthritis.17
Center for Disease Control (CDC) recommends PCV-13 followed by PPSV-23 at least 8 weeks later for general population. For those who have already received PPSV-23, PCV-13 should be given at least 1 year later with and additional PPSV-23 booster given as usual 5 years from the first18
EULAR Guidelinesstrongly recommend pneumococcal vaccination inall patients with rheumatic diseases11.
Table 1: Immunogenicity of various vaccines in the presence of various immunosuppressants in RA and SLE
| Methotrexate | TNFi | Rituximab | Abatacept | Tofacitinib | Tocilizumab | |
|---|---|---|---|---|---|---|
| RA | ||||||
| Influenza | ± | + | ↓↓ | ↓ | + | + |
| Pneumococcal | +* | +* | ↓↓ | ↓ | ↓ | + |
| SLE | ||||||
| Influenza | + | + | ↓↓ | ↓ | NA | NA |
| Pneumococcal | ± | + | ↓↓ | NA | NA | NA |
* combination – reduced immunogenicity; ± Doubtful; ↓ Reduced; + Intact immunogenicity; NA,Not available; TNFi, TNF inhibitor
Hepatitis B
Studies in RA, SLE, Ankylosing Spondylitis, Behcet’s disease suggest immunogenicity of the Hepatitis B vaccination irrespective of disease activity, steroid or DMARD use. However, the amount of the data is insufficient to draw meaningful conclusions. EULAR guidelines recommend Hepatitis B vaccination for the patients at risk including intravenous drug abuse, multiple sex partners in the previous 6 months or health care personnel11. Hepatitis B vaccination is a part of universal immunization programme in India.
Herpes Zoster
Herpes zoster infection risk is increased in Rheumatic diseases. Special concerns regarding Herpes Zoster are being raised in view of increased risk in patients of RA receiving tofacitinib.As HZV is a live attenuated vaccine, its use in immunosuppressed patients is controversial. However, evidence is accumulating from larger registry based studies suggesting its safety in immunosuppressed patients with rheumatic diseases19. American Advisory Committee on Immunization Practices(ACIP) recommends using HZV in general population ≥ 50 years, persons anticipating immunosuppressant (at least two weeks prior to administration of immunosuppressive agent), in persons taking low-dose immunosuppressive therapy (e.g., <20 mg/day of prednisone or equivalent or using inhaled or topical steroids)20. Temporary discontinuation of immunosuppressive medication before vaccination with live attenuated vaccines might also be considered, but there are no studies to support this strategy.
Live vaccination should be avoided in following scenarios
- Steroids - steroid more than 10 mg for two weeks or more
- cDMARDs- Cyclosporine>2.5 mg/kg per day, Sulfasalazine >40 mg/kg per day or 2 g/day, Azathioprine>3 mg/kg,Cyclophosphamide >2.0 mg/kg per day, Leflumomide>0.5 mg/kg per day
- Biologic except B cell depletion therapy (BCDT) - Avoid anti TNF for four weeks
- BCDT - Avoid after BCDT for 6 months and can be given 4 weeks prior to BCDT initiation
Vaccine coverage in an outpatient rheumatology clinic in Germany were 18% and 25% for pneumococcal and influenza respectively.21 Another telephone based survey reported reasons for failure to receive pneumococcal and influenza vaccine were lack of doctor recommendations (55%), safety or efficacy concern (21%) and lack of motivation (19%)22. Simple interventions shown to be useful in increasing coverage include: presentation to rheumatology providers, creation of immunization algorithm, placing reminders on clinic forms, stocking the vaccine in clinic, establishing protocols for vaccination at admission.
To summarize, Box 2 shows the EULAR recommendations for the vaccination of individuals with AIRD. Recently, updates of these guidelines were presented in EULAR Meeting, Amsterdam, 2018.
- Vaccination status should be assessed in the initial work-up of patients
- Vaccine should ideally be administered to patients with an AIRD during stable disease
- Live attenuated vaccines should be avoided whenever possible.
- Vaccine can be administered to patients being treated with DMARDs and TNF inhibitors, but vaccine should be administered before starting B-cell-depleting biologic therapy
- Influenza vaccination should be strongly considered
- PPV23 should be considered
- Patients with an AIRD should have TT vaccination in accordance with the recommendations for the general population; in case of major or contaminated wounds in patients who received rituximab within 24 weeks, tetanus immunoglobulin instead of TT vaccine should be administered
- Herpes zoster vaccination “can” be considered
- For hyposplenic or asplenic patients, influenza, pneumococcal and H. influenzae type b and meningococcal C vaccinations are recommended
- HepatitisA and hepatitis B vaccination are only recommended for patients with an AIRD who are ‘at risk’ (i.e., intravenous drug abuse, multiple sex partners in the previous 6 months, or health care personnel)
- Patients who plan to travel are recommended to have vaccinations according to general rules, except for live-attenuated vaccines, which should be avoided whenever possible by immunosuppressed patients
- BCG vaccination is not recommended
It is under the process of publication. Newer recommendations include: immunocompetent household members of patients with AIIRD should be encouraged to receive vaccines according to national guidelines with the exception of oral poliomyelitis vaccine and live attenuated vaccine should be avoided for the first 6 months in newborn whose mother received biologics in second half of pregnancy.
bDMARDS and Vaccination
- Ideally, vaccination should be given (live or killed) four weeks before starting B cell depletion therapy. However, partial efficacy has been noted when given at least two weeks before Rituximab.
- Killed vaccine can be given during treatment with anti TNF, tocilizumab.
- JAK inhibitor predispose to Herpes Zoster reactivation. Herpes Zoster vaccine should be given at least two weeks before starting JAK inhibitor.
- Live attenuated vaccines should be avoided whenever possible.
Table 2 summarises immunogenicity of vaccines in various CTDs, disease flare and recommendations.
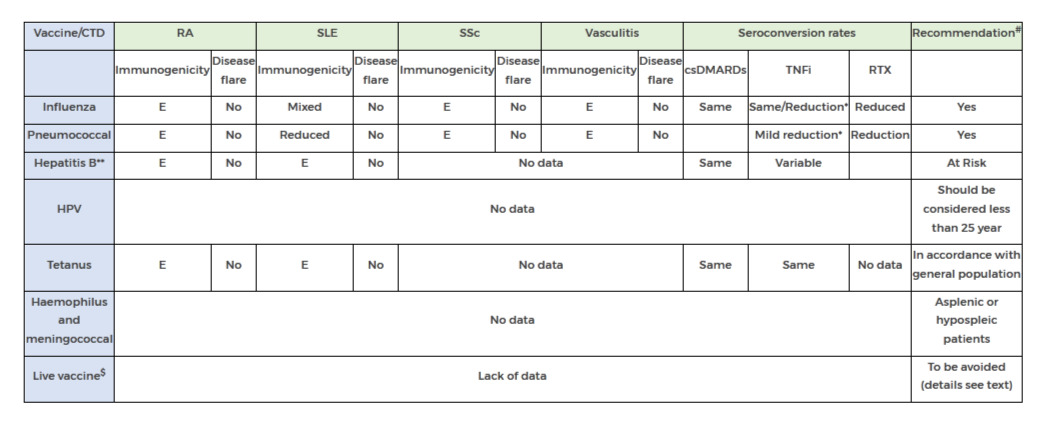
Table2: Efficacy of Vaccine in autoimmune rheumatic disease *with methotrexate; **more data needed; #see text for B cell depletion therapy; $BCG vaccine, oral poliomyelitis vaccine, oral typhoid fever vaccine and yellow fever vaccine csDMARDs, Conventional DMARDs; bDMARDs, biologics; E, Effective;RTX, Rituximab
REFERENCES
- 1. Stojanovich, L. Influenza vaccination of patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). Clin. Dev. Immunol.13, 373–375 (2006).
- 2. Chang, C.-C., Chang, Y.-S., Chen, W.-S., Chen, Y.-H. & Chen, J.-H. Effects of annual influenza vaccination on morbidity and mortality in patients with Systemic Lupus Erythematosus: A Nationwide Cohort Study. Sci. Rep.6, 37817 (2016)..
- 3. Louie, J. S. et al. Clinical and antibody responses after influenza immunization in systemic lupus erythematosus. Ann. Intern. Med.88, 790–2 (1978).
- 4. Holvast, A. et al. Safety and efficacy of influenza vaccination in systemic lupus erythematosus patients with quiescent disease. Ann. Rheum. Dis.65, 913–918 (2005).
- 5. Setti, M. et al. Flu vaccination with a virosomal vaccine does not affect clinical course and immunological parameters in scleroderma patients. Vaccine27, 3367–3372 (2009).
- 6. Holvast, A. et al. Wegener’s granulomatosis patients show an adequate antibody response to influenza vaccination. Ann. Rheum. Dis.68, 873–878 (2009).
- 7. Pasoto, S. G. et al. Short and long-term effects of pandemic unadjuvanted influenza A(H1N1)pdm09 vaccine on clinical manifestations and autoantibody profile in primary Sjögren’s syndrome. Vaccine31, 1793–1798 (2013).
- 8. Westra, J., Rondaan, C., Van Assen, S. & Bijl, M. Vaccination of patients with autoimmune inflammatory rheumatic diseases. Nat. Rev. Rheumatol.11, 135–145 (2015).
- 9. Kapetanovic, M. C. et al. Impact of anti-rheumatic treatment on immunogenicity of pandemic H1N1 influenza vaccine in patients with arthritis. Arthritis Res. Ther.16, R2 (2014).
- 10. Kumar, S., Rath, P. & Malaviya, A. A practical guide to adult vaccination for patients with autoimmune inflammatory rheumatic diseases in India. Indian J. Rheumatol.12, 160 (2017).
- 11. van Assen, S. et al. EULAR recommendations for vaccination in adult patients with autoimmune inflammatory rheumatic diseases. Ann. Rheum. Dis.70, 414–422 (2011).
- 12. Friedman, M. A. & Winthrop, K. Vaccinations for rheumatoid arthritis. Curr. Opin. Rheumatol.28, 330–336 (2016).
- 13. Bingham, C. O. et al. Immunization responses in rheumatoid arthritis patients treated with rituximab: Results from a controlled clinical trial. Arthritis Rheum.62, 64–74 (2010).
- 14. Kapetanovic, M. C. et al. Influence of methotrexate, TNF blockers and prednisolone on antibody responses to pneumococcal polysaccharide vaccine in patients with rheumatoid arthritis. Rheumatology45, 106–111 (2006).
- 15. Mathian, A., Pha, M. & Amoura, Z. Lupus and vaccinations. Curr. Opin. Rheumatol. 1 (2018). doi:10.1097/BOR.0000000000000525
- 16. MERCADO, U., ACOSTA, H. & DIAZ-MOLINA, R. Antibody Response to Pneumococcal Polysaccharide Vaccine in Systemic Sclerosis. J. Rheumatol.36, 1549–1550 (2009).
- 17. Mease, P. J. et al. Pneumococcal vaccine response in psoriatic arthritis patients during treatment with etanercept. J. Rheumatol.31, 1356–61 (2004).
- 18. Kobayashi, M. et al. Intervals Between PCV13 and PPSV23 Vaccines: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR. Morb. Mortal. Wkly. Rep.64, 944–947 (2015).
- 19. Yun, H. et al. Risk of Herpes Zoster in Autoimmune and Inflammatory Diseases: Implications for Vaccination. Arthritis Rheumatol.68, 2328–2337 (2016).
- 20. Dooling, K. L. et al. Recommendations of the Advisory Committee on Immunization Practices for Use of Herpes Zoster Vaccines. MMWR. Morb. Mortal. Wkly. Rep.67, 103–108 (2018).
- 21. Krasselt, M., Baerwald, C. & Seifert, O. Insufficient vaccination rates in patients with systemic lupus erythematosus in a German outpatient clinic. Z. Rheumatol.77, 727–734 (2018).
- 22. Lawson, E. F., Trupin, L., Yelin, E. H. & Yazdany, J. Reasons for failure to receive pneumococcal and influenza vaccinations among immunosuppressed patients with systemic lupus erythematosus. Semin. Arthritis Rheum.44, 666–71 (2015).
Authors
Dr Sakir Ahmed
KIMS, Bhubaneshwar
Ipsita Mohanty
Bhubaneshwar
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma
| DMARD | General Safety recommendations | Dose modificationin renal dysfunction. | Dose modification in liver dysfunction |
|---|---|---|---|
| Methotrexate | Monitor patients closely for bone marrow, liver, lung and kidney toxicities | CrCl 10-50 ml/min: 50% of dose at normal dosing interval CrCl<10 ml/min: avoid use | Bilirubin 3.1-5.0 mg/dl or AST> 3 times ULN: give 75% of dose Bilirubin >5.0 mg/dl: avoid use |
| Leflunomide |
Can cause severe liver injury Recommend ALT monitoring monthly for 6 months after initiating, and q6-8weeks thereafter If ALT rises to >3x ULN, interrupt therapy while investigating probable cause; if likely leflunomide-induced, initiate cholestyramine washout to speed elimination and conduct follow-up LFTs at least weekly until ALT value within normal range; if not leflunomide-induced ALT elevation, may consider resuming leflunomide |
There are no dosage adjustments provided in the manufacturer’s labelling | Not recommended for use in patients with pre-existing liver disease or those with baseline ALT>2 times ULN; monitor liver function closely. Use is contraindicated in hepatic impairment. |
| Sulfasalazine |
Can lead to hepatobiliary disorders: reports of hepatotoxicity, including elevated liver function tests cholestatic jaundice, cirrhosis, hepatitis cholestatic, cholestasis and possible hepatocellular damage including liver necrosis and liver failure; Renal and urinary disorders: nephrolithiasis reported |
Renal clearance: 37% There are no dosage adjustments provided in the manufacturer's labelling; use with extreme caution |
Data not available |
| Hydroxychloroquine | Both chloroquine and HCQ can cause a 10 percent decrease in creatinine clearance by competitively inhibiting creatinine secretion; this does not represent a true change in renal function. |
Excretion of these drugs is principally by direct renal clearance of the parent compound and hepatic metabolites. Manufacture does not provide instructions for use in renal failure. Expert recommendation is reduction of dose <250mg/day. antimalarials have been found in the urine five years after medication was stopped |
Data not available. Dose should be reduced if continued. |
| Azathioprine | Increased risk of infection and hepatotoxicity; monitor liver function periodically; hepatic sinusoidal obstruction syndrome reported; discontinue therapy if suspected |
CrCl>50 ml/minute: no adjustment recommended. CrCl 10 to 50 ml/minute: administer 75% of normal dose. CrCl<10 ml/minute: administer 50% of normal dose. Haemodialysis (partially dialyzable; ~45% removed in 8 hours): administer 50% of normal dose; supplement: 0.25 mg/kg. CRRT: administer 75% of normal dose. |
There are no dosage adjustments provided in the manufacturer’s labelling. However expert recommendation is that it may be used with caution. |
| Mycophenolate mofetil | Toxicity may increase in renal impairment; use caution |
there have been no specific dosage adjustments identified, Although use of lower doses may be required. Mycophenolic acid (MPA) exposure appears to be inversely related to renal function. With GFR less than 25mL/min/1.73m2 in renal transplant recipients doses more than 2g/d should be avoided. |
however, it is not currently known whether dosage adjustments are necessary for hepatic disease with other aetiologies. Increased monitoring may be necessary in patients with hyperbilirubinemia and/or hypoalbuminemia |
| Apremilast | Renal/hepatic impairment |
Severe renal impairment (CrCl<30 ml/min): reduce dose to 30 mg po qday Mild-to-moderate renal impairment: no dosage adjustment required |
Hepatic impairment: no dosage adjustment required |
| Tacrolimus |
Increased mortality in female liver transplant patients. Renal impairment does not affect the elimination or serum concentrations of tacrolimus; however, tacrolimus may cause nephrotoxicity requiring dose reduction. Post-transplant hepatic impairment may be associated with increased risk of developing renal insufficiency |
Use lower end of dosing range Monitor renal function and adjust dose according to whole blood concentrations and tolerability |
Mild: no dosage adjustment required Moderate: monitor whole blood concentrations and adjust dose accordingly Severe (mean child-pugh score >10): mean clearance of tacrolimus was substantially lower compared with normal hepatic function; dosage reduction recommended; administer 80% of preconversion daily dose of immediate release dosage form when converting from tacrolimus immediate release to extended release |
| Cyclophosphamide | Use with caution in patients with hepatic or renal impairment | Renal impairment: CrCl<10 ml/min, give 75% of normal dose; CrCl>10 ml/min, give full dose | Hepatic impairment: give 75% of normal dose if transaminase levels are >3 times upper limit of normal or bilirubin is 3.1-5 mg/dl |
| Rituximab | Increased risk of potentially fatal hepatitis b virus reactivation | There are no dosage adjustments provided in the manufacturer's labelling (has not been studied) | There are no dosage adjustments provided in the manufacturer's labelling (has not been studied) |
|
Infliximab Adalimumab Etanercept Golimumab |
There are no dosage adjustments provided in the manufacturer's labelling. There are case reports of successful use in renal or hepatic failure. | ||
| Secukinumab | Data not available | Data not available | Data not available |
| Tofacitinib | Associated with increased LFTs |
Mild: no dosage adjustment required RA or PsA Moderate-to-severe: not to exceed 5 mg qday UC Moderate-to-severe: if taking 10 mg bid, reduce to 5 mg bid; if taking 5 mg bid, reduce to 5 mg qday |
Mild: no dosage adjustment required Severe: not recommended Ra or PsA Moderate-to-severe : not to exceed 5 mg qday UC Moderate-to-severe: if taking 10 mg bid, reduce to 5 mg bid; if taking 5 mg bid, reduce to 5 mg qday |
| Baricitinib | Data not available |
Renal impairment Egfr ≥60 ml/min/1.73 m²: renal function significantly affects Baricitinib systemic exposure; monitor closely eGFR<60 ml/min/1.73 m²: not recommended |
Hepatic impairment Mild or moderate: no dose adjustment required Severe: not recommended |
Authors
Dr Sakir Ahmed
KIMS, Bhubaneshwar
Ipsita Mohanty
Bhubaneshwar
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma
Reviewed and Edited by : Dr Avinash Jain
This document is a summary of guidelines of EULAR, ACR and BSR along with the inputs from a literature search. It is designed to be brief and in simple language understandable both to the patient and the specialist. However patients are strongly advised not to interpret the drug advisory on their own without consulting their treating physicians.
A. A successful pregnancy is possible in almost all rheumatic diseases provided disease is well-controlled, and there is no permanent organ damage.
- 1. Pregnancies are more likely to be successful when they are planned, with adequate discussion among the patient, the rheumatologist and the obstetrician.
- 2. Successful, however, does not mean uneventful. Doctors and patients must be prepared to deal with possible complications for both mother and child.
- 3. Diseases with the potential to affect the kidneys, or lung (including increasing pressure of the pulmonary arteries) like lupus, antiphospholipid syndrome, inflammatory myositis, systemic sclerosis and overlap syndromes are more likely to affect pregnancy outcome than others.
- 4. Persistently raised creatinine (end-stage renal disease) or high pulmonary arterial pressures may hinder successful pregnancies. In fact in these conditions, pregnancy may worsen the health condition of the mother.
- 5. Any rheumatic diseasemust be under optimal control for 6 months before pregnancy is planned.
B. Effects of rheumatic diseases on pregnancies:
- 1. In the absence of permanent organ damage, the fertility of patients is not altered due to rheumatic diseases. Diseases like systemic sclerosis or sjogrensyndrome may lead to dyspareunia.
- 2. Uncontrolled rheumatic diseases have a lot of inflammation and may lead to pregnancy loss especially in the 1st
- 3. In the later phase there are more chances of pregnancy induced hypertension and foetal growth retardation. Patients who have or have had kidney disease, due to vasculitis, scleroderma, or lupus, generally have an increased risk of severe hypertension and pre-eclampsia.
- 4. Pulmonary arterial hypertension worsens in the post-partumperiod.Patients with high pulmonary artery pressures are advised not to get pregnant.
- 5. APS probably has the greatest impact on pregnancy. It causes both early and late miscarriage.Other complications include premature birth, low-weight babies, athrombosis (condition where blood clots form in the blood vessels) and pre-eclampsia. Thus, pregnancy with APS should always be considered as high risk and require close medical and obstetric monitoring. Treatment is based on low-dose aspirin and heparin.
- 6. Babies of mothers having anti-Ro antibodies (in Sjogren or lupus) are at higher risk of developing congenital heart blocks. The anti-Ro antibodies may interfere with the development of the electric conduction system of the heart. Thus mother with anti-Ro antibodies needsfoetal heart monitoring with foetal echocardiography (ultrasound of foetalheart) during 2nd
- 7. It is important to discuss the possible effects of various anti-rheumatic drugs on pregnancy. There should be expert assessment of risk-benefit to determine the drugs to be continued during pregnancy starting from the pre-conception stage.
C. Effect of pregnancies on rheumatic diseases:
- The earlier paradigm was that diseases like rheumatoid arthritis tend to go into quiescence (2/3rd of RA) while diseases like lupus would invariably flare (50% of SLE flare with 20% being major organ flares) during pregnancy.
- With newer treatment strategies and better disease control, studies have shown that only a minority (~10%) of RA have improved disease activity while lupus patients (in remission for at least 6 months, and on hydroxychloroquine) do not have increased flare rates.
- It is very important to report any new symptoms and any worsening to both your rheumatologist and obstetrician.
D. Drugs permissible in pregnancy:
- Previously drugs were prescribed followingUSFDA pregnancy categories: A, B, C, D, and X. However these are not water-tight compartments and this has lead the FDA to abandonthis approach. Thus, risk-benefit of each drugs should be discussed in context of the patient, the disease and the age of gestation.
- As overarching guidelines, we endorse the ACR recommendation that currently stand as: (please also see the BSR guidelines: table 2)
| Pregnancy | Lactation | ||
|---|---|---|---|
| NSAID | Yes (avoid after 32 weeks) | Yes | |
| Sulfasalazine | Yes | Yes | |
| Antimalarials | Yes | Yes | |
| Corticosteroids | Yes | Yes | |
| Cyclosporine/Tacrolimus | Yes | Probably yes | |
| Azathioprine | Yes | Probably yes | |
| Mycophenolate | No | No | |
| Methotrexate | No | No | |
| Cyclophosphamide | No | No | |
| Anti-tumor necrosis factor (TNF) | Yes | Yes | |
| Rituximab | No | No | |
| Warfarin | No (with caution, only after first trimester) | Yes | |
| Heparin | Yes | Yes | |
| Table 1: Acceptable medications during pregnancy and lactation | |||
- There is strong evidence that hydroxychloroquine must be continued in lupus and APS during pregnancy and it is our personal opinion that it should be continued in RA during pregnancy as well.
- Captopril and enalapril are to be avoided in pregnancy but are safe drugs during breastfeeding.
- Other drugs:
Tocilizumab
- Tocilizumab (TCZ) should be stopped at least 3 months before conception, but unintentional exposure early in the first trimester is unlikely to be harmful
- There are no data upon TCZ use in breastfeeding
Abatacept
- There are insufficient data to recommend abatacept (ABA) in pregnancy. Unintentional exposure early in the first trimester is unlikely to be harmful
- There are no data upon ABA use in breastfeeding
Secukinumab
- There are insufficient data to recommend Secukinumab(SEK) in pregnancy.
- There are no data upon SEK use in breastfeeding
Preconception planning for males:
- Limited data is available but it has shown that Methotrexate is unlikely to affect male fertility (table 2). Currently there are no recommendations to stop it during pre-conception stage.
- For sulfasalazine, conception rates may be enhanced by stopping sulfasalazine for 3 months prior to conception
- Thalidomide should be stopped at least 3 months in advance prior to planned conception (as it is present in spermatozoa).
Summary::
- The likelihood of a successful and healthy pregnancy is highest if kidney and heart function, and blood pressure are normal.
- The disease is inactive for at least 6 months prior to conception.
- Drugs should be used after patient tailored risk-benefit assessment.
Table 2: BSR guideline for drugs used in rheumatology during pregnancy:
| Summary of drug compatibility in pregnancy and breastfeeding | |||||
|---|---|---|---|---|---|
| Compatible peri-conception | Compatible with first trimester | Compatible with second/third trimester | Compatible with breastfeeding | Compatible with paternal exposure | |
| CORTICOSTEROIDS | |||||
| Prednisolone | Yes | Yes | Yes | Yes | Yes |
| Methylprednisolone | Yes | Yes | Yes | Yes | Yes |
| ANTIMALARIALS | |||||
| Hydroxychloroquine | Yes | Yes | Yes | Yes | Yes* |
| ANTIMALARIALS | |||||
| DMARDS | |||||
| Methotrexate <20mg/week | Stop 3 months in advance | No | No | No | Yes* |
| Sulfasalazine (with 5mg folic acid) | Yes | Yes | Yes | Yes† | Yes† |
| Leflunomide | Cholestyramine washout, no | No | No | No Data | Yes* |
| Azathioprine <2mg/kg/day | Yes | Yes | Yes | Yes | Yes |
| Ciclosporin | Yes | Yes§ | Yes§ | Yes* | Yes* |
| Tacrolimus | Yes | Yes§ | Yes§ | Yes* | Yes* |
| Cyclophosphamide | No | No| | No| | No | No |
| Mycophenolate mofetil | Stop 6 weeks in advance | No | No | No | Yes* |
| Intravenous immunoglobulin | Yes | Yes| | Yes | Yes | Yes* |
| ANTI-TNF | |||||
| Infliximab | Yes | Yes | Stop at 16 weeks | Yes* | Yes* |
| Etanercept | Yes | Yes | Second but not third | Yes* | Yes* |
| Adalimumab | Yes | Yes | Second but not third | Yes* | Yes* |
| Certolizumab | Yes | Yes | Yes* | Yes* | No data |
| Golimumab | No data | No data | No data | No data | No data |
| Cyclophosphamide | No | No| | No| | No | No |
| Cyclophosphamide | No | No| | No| | No | No |
| OTHER BIOLOGICS | |||||
| Rituximab | Stop 6 months in advance | No¶ | No | No data | Yes* |
| Tocilizumab | Stop 3 months in advance | No¶ | No | No data | No data** |
| Anakinra | No | No¶ | No | No data | No data** |
| Abatacept | No | No¶ | No | No data | No data** |
| Belimumab | No | No¶ | No | No data | No data** |
| CONVENTIONAL PAINKILLERS | |||||
| Paracetamol | Yes | Yes†† | Yes†† | Yes | Yes‡‡ |
| Codeine | Yes | Yes | Yes | Caution | Yes‡‡ |
| Tramadol | Yes | Yes | Yes | Yes§§ | Yes‡‡ |
| OTHER CHRONIC PAIN TREATMENTS | |||||
| Amitriptyline | Yes | Yes | Yes | Yes | Yes‡‡ |
| Gabapentin | No | Insufficient data|| | Insufficient data|| | Insufficient data | No data |
| Pregabalin | No data | No data | No data | No data | No data |
| Venlafaxine | Yes | Yes | Yes | Insufficient data|| | Yes‡‡ |
| Fluoxetine | Yes | Yes | Yes | Caution|| | Yes‡‡ |
| Paroxetine | Yes | Yes | Yes | ICaution|| | Yes‡‡ |
| Sertraline | Yes | Yes | Yes | ICaution|| | Yes‡‡ |
| NSAIDS | |||||
| NSAIDs | Yes | Caution¶¶ | Stop by week 32 | Yes | Yes |
| COX-2 inhibitors | No | No | No | No | No data |
| Low-dose aspirin | Yes | Yes | Yes | Yes*** | Yes‡‡ |
| ANTICOAGULANTS | |||||
| Warfarin | No | No | No/Caution | Yes | No data |
| Low-molecular-weight heparin | Yes | Yes | Yes | Yes*** | Yes‡‡ |
| Dabigatran | No data | No data | No data | No data | No data |
| BISPHOSPHONATES | |||||
| Bisphosphonates | Stop 6 months in advance | No | No | No data | No data |
| ANTIHYPERTENSIVES | |||||
| Angiotensin-converting-enzyme inhibitor | Stop when pregnancy confirmed | No | Yes§§ | No data | |
| Nifedipine | Yes | Yes<60mg/day | Yes<60mg/day | Yes | Yes‡‡ |
| Amlodipine | No data | No data | No data | No data | Yes*** |
| PULMONARY VASODILATORS | |||||
| Sildenafil | No data | No data | No data | No data | No data |
| Bosentan | No data | No data | No data | No data | No data |
| Prostacyclin | No data | No data | No data | No data | No data |
|
NSAIDS=non-steroidal anti-inflammatory drugs; COX-2=cyclooxygenase-2; MDT=multidisciplinary team. * Data are limited † In healthy full-term infants only ‡ Conception may be enhanced by stopping sulfasalazine for 3 months prior to conception § Suggested monitoring of maternal blood pressure, renal function, blood glucose and drug levels | Only consider in severe or life-/organ-threatening maternal disease ¶ Unintentional first trimester exposure is unlikely to be harmful ** Unlikely to be harmful †† Intermittent use advised, see full guideline for details ‡‡ No studies identified, but unlikely to be harmful due to maternal compatibility §§ Limited evidence, but unlikely to be harmful || Insufficient evidence regarding use for treatment of chronic pain in pregnancy ¶¶ Possible association with miscarriage and malformation *** No studies identified, but unlikely to be harmful. |
|||||
Table 3 The impact of rheumatic disease on pregnancy and vice-versa.
| Rheumatic Diseases | Impact of pregnancy on rheumatic disease |
Pregnancy outcome > IUGR/premature/SGA |
Foetal Loss |
Other complications |
Postpartum | Fertility |
|---|---|---|---|---|---|---|
| RA | Decreased disease activity (found in only a minority of recent studies under T2T regimens) | Patients on glucocorticoids maybeat risk for small for gestational age and for preterm delivery | not been convincingly shown to be associated with an increase in foetal morbidity or foetal losses | pregnancy outcomes in women with well-controlled RA are comparable to those in the general population | Flares in up to 90%.Usually not in well controlled disease. | Preserved. |
| SLE | Increased flare rates. Patients on HCQ have similar flare rates as non-pregnant counterparts | extremely variable rate of induced abortions reported. Possibility of CHB if anti-Ro present in mother. Concomitant APS increases risk | ~5% | two- to fourfold increased rate of obstetric complications including preterm labour, unplanned caesarean delivery, foetal growth restriction, preeclampsia, and eclampsia. Patients with SLE also have significantly higher risk of thrombosis, infection, thrombocytopenia, and transfusion | Flares. Usually not in well controlled disease. | Preserved in well controlled lupus without organ damage. High dose CYC is a risk factor for reduced fertility. Apparent infertility is common due to effects on their own self-esteem and mental well-being, and stress with partner. |
| APS | Potentially increased risks of thrombosis especially in post-partum period | Up to 50% of treated cases have PIH and related foetal complications | up to 80% risk of current pregnancy loss without treatment. 20% with treatment | Operative deliveries are commoner; increased risk of PIH, placental insufficiency and abruption, HELLP syndrome and pre-term labour. Rarely foetal thrombosis | Increased risk of thrombosis | Much lowered |
| SSc | Less data available. May exacerbate vasculopathy like PAH, raynaud, or risk for renal crisis | Limited data: possibly more premature births and more infants small for gestational age | Multiple studies (but not all) suggest increased risk of abortion; but most have small numbers | increased frequency of preterm delivery, intrauterine growth restriction, and low-birthweight baby | Data is less but apparently fertility is maintained. Dyspareunia can be an issue | |
| Sjogren | Like lupus: likely to worsen during pregnancy and more so in the postpartum period, especially in presence of PAH | Increased risk of foetal growth restriction. | variable rate of induced abortions |
prevalence of CHB is 1-2%. Recurrence rates are 10-20%. Neonatal lupus risk is ~2% |
Flares. Flares expected to be less in well controlled disease |
Data is less but apparently fertility is maintained in absence of organ damage. Dyspareunia can be an issue |
| Takayasu | Unknown; theoretically possible to increase long term morbidity | ~25% have growth retardation; | 25% | Operative deliveries are commoner (~40%); increased risk of PIH and pre-term labour | Unknown | Data is less. Major determinants of fertility are hypertension, cardiac involvement and renal (artery) involvement |
| ANCA associated vasculitis | Data is very limited but around 20% flare during pregnancy | Limited data | 10% of cases in GPA, up to 20% in EGPA (under optimal conditions) | 20% preterm | Unknown | Decreased |
T2T: treat to target strategy; PAH: pulmonary arterial hypertension; HCQ: hydroxychloroquine; ANCA: anti-neutrophil cytoplasmic antibodies; CHB: complete heart block
Click to downloadAuthor
Dr Avinash Jain
SGPGI, Lucknow
Reviewed by : Dr Liza Rajshekhar, Dr Vikas Agarwal, Dr Aman Sharma, Dr Praveen Hisaria and Dr Sapan C Pandya
This is an area which is often neglected and is a burden too heavy to be carried by the weakened bones! There is an increased risk of osteoporosis (OP) in rheumatic diseases, a chronic inflammatory state, often warranting the use of steroids. Pathogenesis is multifactorial involving cross-talk between inflammatory cells and bone cells, disease complications, poor nutrition, medications, and decreased physical activity. Dickkopf-related protein 1 (DKK-1) and sclerostin, which are negative regulators of the Wntsignalling pathway, inhibit bone formation in rheumatic diseases.[1] Steroid use increases their expression besides augmenting osteoclastogenesis by inhibiting Osteoprotegerin (OPG) and increasing RANKL expression. Muscle wasting and changes in bone microstructure further compound the problem. Factors which increase the risk of OP have been outlined in Box
1.OP screening involves
- 1. A good history and examination to asses risk factors and h/o fracture and examination including BMI and loss of height, reduced space between lower ribs and pelvis, spinal tenderness.
- 2. Look for secondary causes
- 3. Addictions particularly tobacco use
- 4. Medications
- 5. Biochemistry including calcium, ALP
- 6. Bone mineral density (BMD) assessment within six months of initiation of glucocorticoid treatment. Most widely used tool is Dual-energy x-ray absorptiometry (DXA) thought newer techniques like pDXA (peripheral DXA), Quantitative Computerised Tomography (QCT) and peripheral QCT are available but needs validation and are costlier.
- 7. Biochemical biomarkers like C telopeptide, free deoxypyridinoline may independently predict fracture risk but are not routinely used
Box 1: Risk Factors
- 1. Age
- 2. Previous h/o low trauma fractures
- 3. Low BMI, Significant weight loss
- 4. Current smoking and alcohol
- 5. Parental h/o hip fracture
- 6. Use of steroids
- 7. Rheumatic diseases
- 8. Secondary OP - Endocrine causes, IBD, malabsorption, Chronic liver disease
- 9. Malnutrition
Fracture Risk Assessment Tool (FRAX) developed by University of Sheffield, estimates 10- year probability of hip and major OP fracture (hip, clinical spine, proximal humerus, or forearm) between 40-90 years using clinical risk factors with/without femoral neck BMD.[2] Besides age, gender, weight, height, it includes risk factors as defined in Box 1 where only Rheumatoid Arthritis is taken in rheumatic diseases, steroid use has been defined as ≥ 5mg for 3 or more months. There are however insufficient data to develop prediction tools for younger adults and children.
Although the greatest relative risk of fracture is in individuals with osteoporosis(OP), the absolute number of fractures in those with BMD T-scores in the low bone mass (osteopenia) range is the same or greater than in those with T-scores in the osteoporosis range, as more individuals belong to the latter category. At times BMD may give us a fall sense of hope and some patients who need preventive therapy may be missed.
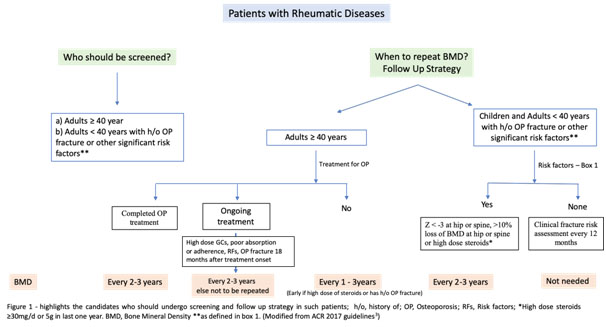
Rheumatic Diseases
Various studies have lookedat the prevalence of osteoporosis in rheumatic disease. [4,5] Prevalence and some of the associated risk factors have been mentioned in Table 1.
Table 1: Prevalence and Risk Factors of OP in rheumatic diseases[4,5]
| Osteoporosis | Additional Risk Factors | |
|---|---|---|
| SLE | 1.4 – 68% | Reduced sun exposure, high falls, renal involvement, longer disease duration No relation to disease activity |
| RA | 18 – 56% | Longer disease duration, Disease activity, HAQ, RF/ACPA, High CRP |
| AS | 19 – 62% | Older age, long-standing disease, syndesmophyte formation, associated IBD |
| PsA | 11 – 47% | Association with disease duration is controversial |
| SSc | 3 – 51% | Intestinal malabsorption, renal disease, subcutaneous calcinosis, no difference in lcSSc and dcSSc |
| IIM | 25% | Correlation with disease activity unclear |
| JIA | 40-52% of adult patients with JIA | Disease Activity and duration |
It is important to remember that osteoarthritis, syndesmophytes, new bone formation, atherosclerosis may falsely increase BMDmeasurement.[6,7]Management strategy should include FRAX score calculation and risk stratification and the use of OP medications accordingly. Moderate to highrisk patients should be treated with OP medications even if steroids are not used. (Table 2 and Figure 1)
GIOP (Glucocorticoid induced Osteoporosis)
Long-term glucocorticoid therapy causes osteoporotic fractures in about 10-12% of treated adult patients and 30–40% of them have radiographic evidence of vertebral fractures,
in view of higher effects of steroids on trabecular bone[8,9]Daily doses of ≤5 mg prednisolone have been shown to increase fracture risk by ~20%, rising to 60% for doses of ≥20 mg per day.[7,10]Fracture risk is highest particularly within first the 3-6 months and correlates with cumulative dose and daily dose. [10] Still the preventive care is suboptimal and less than a quarter undergo OP assessment and often the therapy is instituted once a fracture occurs.
Whom to Treat?
In GIOP, the terms ‘prevention’ and ‘treatment’ distinguish between the initiation of anti-osteoporosis intervention at the start of glucocorticoid therapy or after >3 months, respectively.
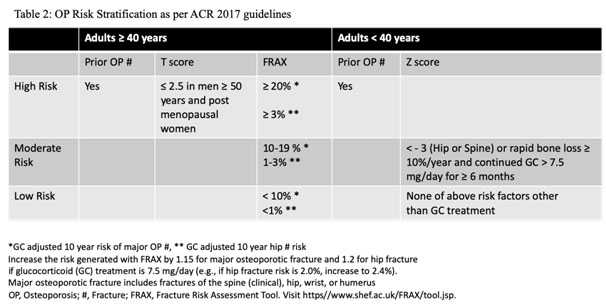
Prevention:
Depending on risk stratification, all adults irrespective of age, not of childbearing age or childbearing age but not planning a pregnancy during treatment, with moderate to high risk as defined in Table 2, should receive oral Bisphosphonate. Second line therapies include IV bisphosphonate, teriparatide, denosumab and raloxifene(for post-menopausal women only) in that order except in women of childbearing potential where teriparatide is preferred over oral bisphosphonate. IV bisphosphonate and denosumab lack safety data in pregnancy and hence should be used only in high risk patients whereas raloxifeneis not recommended. Some authorities do no rate bisphosphonates over teriparatide due to lack of head to head comparisons. Denosumab, though not a first line therapy, but is a good alternative particularly in patients with renal failure. Some experts recommend upfront teriparatide in patients with T-score < -3.5 or T-score of -2.5 or below plus a fragility fracture.Romosozumab, sclerostin inhibitor, is another new emerging therapy yet to find a place in guidelines.Refer to table 3 for dosing, side effects and monitoring.
Otheressential recommendations including for those with low risk of fracture are
- 1. Dailycalcium and vitamin D 1,000–1,200 mg and 600–800 IU respectively
- 2. Lifestyle modifications like cessation of smoking, limiting alcohol intake to 1-2/day,
- 3. Balanced diet and weight and daily exercises.
- 4. Figure 1summarises the prevention strategy and follow up treatment[11]In case of moderate to high risk, continue oral Bisphosphonate for ten years and Zoledronic acid for five years. There is no concept of drug holiday in such cases. Besides GCs, Methotrexate, Cyclophosphamide, heparin, oral anticoagulants, anti-convulsant are some of the medications which have been implicated in OP
Table 2
| Drug | Dose | Pre-requisites or monitoring | Side effects and Contra-indications |
|---|---|---|---|
| Oral Bisphosphonate | Alendronate 70mg/week Risedronate 35mg/week or 150mg/month |
Avoid oral calcium supplements, antacids, magnesium supplements/laxatives, and iron preparations for at least 60 minutes after drug administration Check serum calcium |
GERD/Reflux oesophagitis Osteonecrosis of Jaw (ONJ) - 1 in 10,000 to 1 in 100,000 patient-years Atypical Femur Fracture(AFF) Hypersensitivity GFR < 30ml/min Hypocalcemia achalasia, esophageal stricture, esophageal varices, Barrett's esophagus) or with an inability to follow the dosing requirements (eg, stay upright for at least 30 minutes) Pregnancy and breastfeeding |
| IV Bisphosphonate | Zoledronic 4mg every year | Can have flu like symptoms, give Paracetamol before IV administration Check calcium and correct before drug administration |
Transient hypocalcemia,
Flu like symptoms within 24-72 hours of infusion; Treat with Paracetamol or NSAIDs ONJ, AFF Hypersensitivity GFR < 30ml/min Hypocalcemia Pregnancy and breastfeeding |
| Teriparatide | 20 mcg s.c every day upto 2 years | Hypercalcemia, Dizziness, arthralgia, rhinitis Osteosarcoma Hypersensitivity, hypercalcemia and related conditions |
|
| Denosumab | 60mg s.c every six months | Check calcium and vitamin D and in deficient – correct same before administering Denosumab months Check calcium and vitamin D and in deficient – correct same before administering Denosumab Higher risk in CKD pts, eGFR < 30 and in those with malabsorption and hence more prone to hypocalcemia. Check calcium level 10 days after administration of Denosumab |
Infections Skin Rash Immediate risk of fractures on stopping the drug – add bisphosphonate if denosumab needs to be stopped Hypersensitivity Hypocalcemia |
| Oral Bisphosphonate | Alendronate 70mg/week Risedronate 35mg/week or 150mg/month |
void oral calcium supplements, antacids, magnesium supplements/laxatives, and iron preparations for at least 60 minutes after drug administration Check serum calcium |
GERD/Reflux oesophagitis Osteonecrosis of Jaw (ONJ) - 1 in 10,000 to 1 in 100,000 patient-years Atypical Femur Fracture(AFF) Hypersensitivity GFR < 30ml/min Hypocalcemia achalasia, esophageal stricture, esophageal varices, Barrett's esophagus) or with an inability to follow the dosing requirements (eg, stay upright for at least 30 minutes) Pregnancy and breastfeeding |
| IV Bisphosphonate | Zoledronic 4mg every year | Can have flu like symptoms, give Paracetamol before IV administration Check calcium and correct before drug administration |
Transient hypocalcemia, Flu like symptoms within 24-72 hours of infusion; Treat with Paracetamol or NSAIDs ONJ, AFF Hypersensitivity GFR < 30ml/min Hypocalcemia Pregnancy and breastfeeding |
| Teriparatide | 20 mcg s.c every day upto 2 years | Hypercalcemia, Dizziness, arthralgia, rhinitis Osteosarcoma Hypersensitivity, hypercalcemia and related conditions |
|
| Denosumab | 60mg s.c every six months | Check calcium and vitamin D and in deficient – correct same before administering Denosumab Higher risk in CKD pts, eGFR < 30 and in those with malabsorption and hence more prone to hypocalcemia. Check calcium level 10 days after administration of Denosumab |
Infections Skin Rash Immediate risk of fractures on stopping the drug – add bisphosphonate if denosumab needs to be stopped Hypersensitivity Hypocalcemia |
CKD, Chronic Kidney Disease; GERD, Gastro-esophageal reflux disease; GFR, Glomerular filtration rate;s.c, subcutaneous
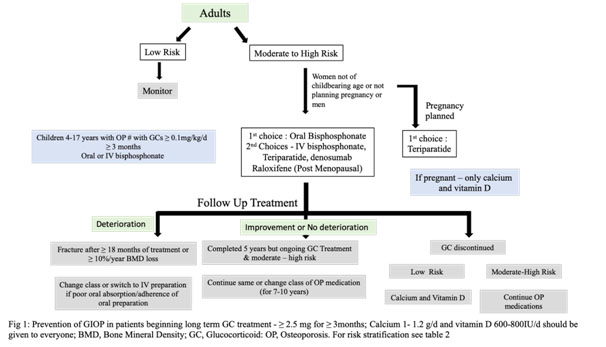
Osteoporosis is a potentially preventable complication which needs to be taken care of by a rheumatologist considering an increasing prevalence of OP in rheumatic diseases. Good control of disease and hence inflammation, minimising steroid use, emphasis on dietary and lifestyle modifications, calcium and vitamin D supplementation and use of OP medications when indicated will help in reducing this complication. “Prevention is always better than cure!Action is better than procrastination.”
Summary
Osteoporosis is a common but neglected issue, amenable to therapy or prevention in patients with rheumatic diseases particularly on steroids
- 1. 10-12% patients on steroid suffer from fracture or 30-50% have radiological evidence of vertebral fracture with maximum loss occurring in 3-6 months.
- 2. Do not forget secondary causes which further increase the risk of osteoporosis.
- 3. All patients with rheumatic diseases, age 40 years or more, and children with risk factors should undergo BMD within six months of initiation of glucocorticoid.
- 4. FRAX score, with correction for steroid dose, can be used in patients aged³ 40 years for fracture risk prediction with/without BMD. (https//www.shef.ac.uk/FRAX/tool.jsp)
- 5. Treatment should be started for those falling in moderate to high risk.
- 6. There is no role of follow up BMD adults population on treatment unless there are risk factors and/or development of fracture despite ³ 18 months of therapy or poor adherence/compliance.
- 7. There is no concept of drug holiday at end of 3-5 years if there is moderate to high risk and therapy should be continued for additional 3-5 years.
- 8. Upfront Teriparatide maybe considered in T-score < -3.5 or T-score of -2.5 or below plus a fragility fracture or a contra-indication for bisphosphonate except pregnancy
References
- 1. Rizzoli R, Biver E. Glucocorticoid-induced osteoporosis: Who to treat with what agent? Nat Rev Rheumatol 2015;11(2):98–109.
- 2. Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008;19(4):385–97.
- 3. Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE, et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis Rheumatol 2017;69(8):1521–37.
- 4. Maruotti N, Corrado A, Cantatore FP. Osteoporosis and rheumatic diseases. Reumatismo 2014;66(2):125
- 5. Gao L-X. Osteoporosis in rheumatic diseases. World J Rheumatol 2015;5(1):14–23.
- 6. van der Weijden MAC, Claushuis TAM, Nazari T, Lems WF, Dijkmans BAC, van der Horst-Bruinsma IE. High prevalence of low bone mineral density in patients within 10 years of onset of ankylosing spondylitis: a systematic review. Clin Rheumatol 2012;31(11):1529–35.
- 7. Staa TP Van, Geusens P, Bijlsma JWJ, Leufkens HGM, Cooper C. Clinical assessment of the long-term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum 2006;54(10):3104–12.
- 8. Curtis JR, Westfall AO, Allison J, Bijlsma JW, Freeman A, George V, et al. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum 2006;55(3):420–6.
- 9. Angeli A, Guglielmi G, Dovio A, Capelli G, de Feo D, Giannini S, et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: A cross-sectional outpatient study. Bone 2006;39(2):253–9.
- 10. van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000;39(12):1383–9.
- 11. Goodman SM, Springer B, Guyatt G, Abdel MP, Dasa V, George M, et al. 2017 American College of Rheumatology/American Association of Hip and Knee Surgeons Guideline for the Perioperative Management of Antirheumatic Medication in Patients With Rheumatic Diseases Undergoing Elective Total Hip or Total Knee Arthroplasty. Arthritis Care Res (Hoboken) 2017;69(8):1111–24.











